
Enhancement of Herpes Simplex Virus (HSV) Infection by Seminal Plasma and Semen Amyloids Implicates a New Target for the Prevention of HSV Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. SEM and SEVI Amyloids and SP Interacted with the HSV Viral Particles
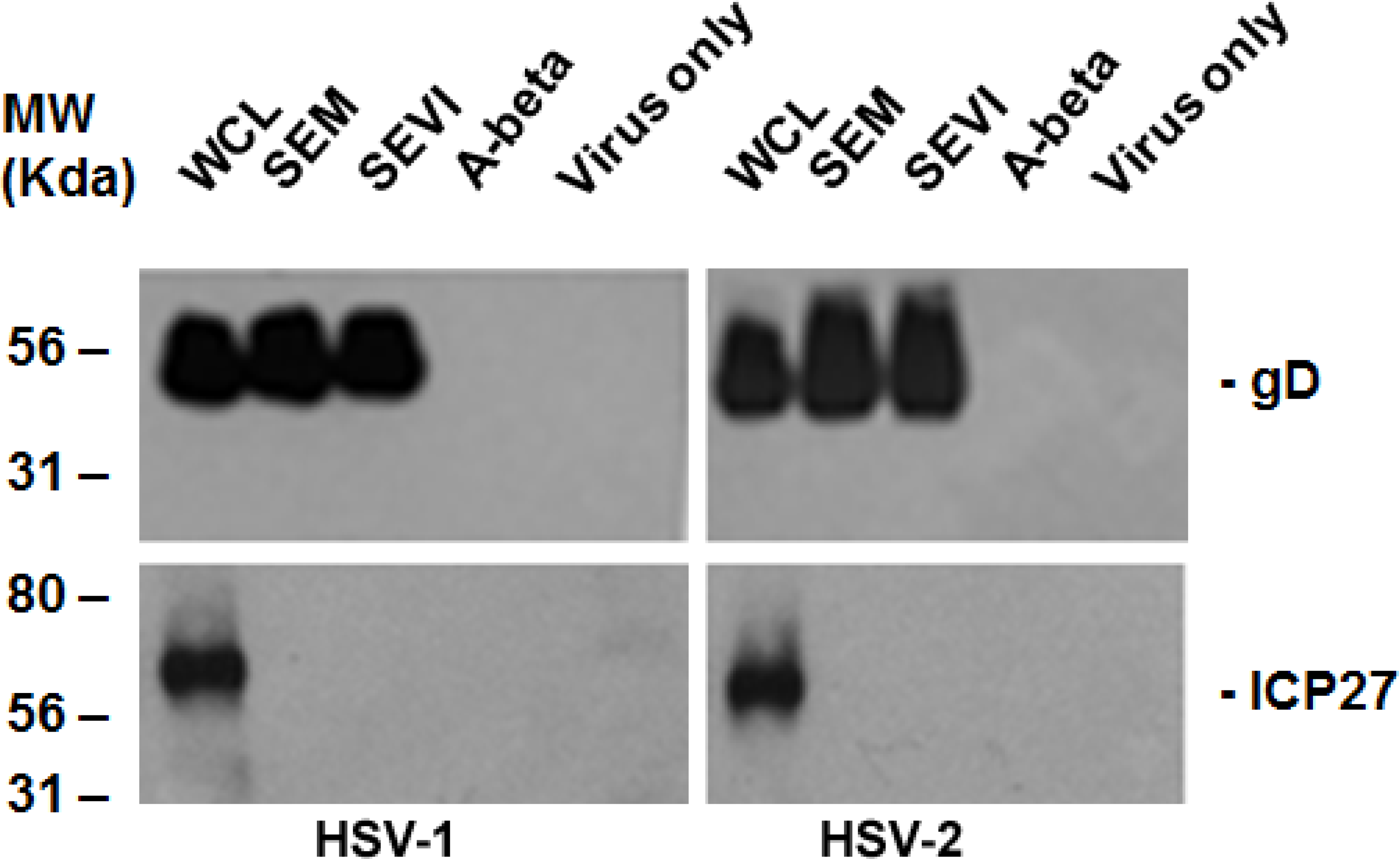
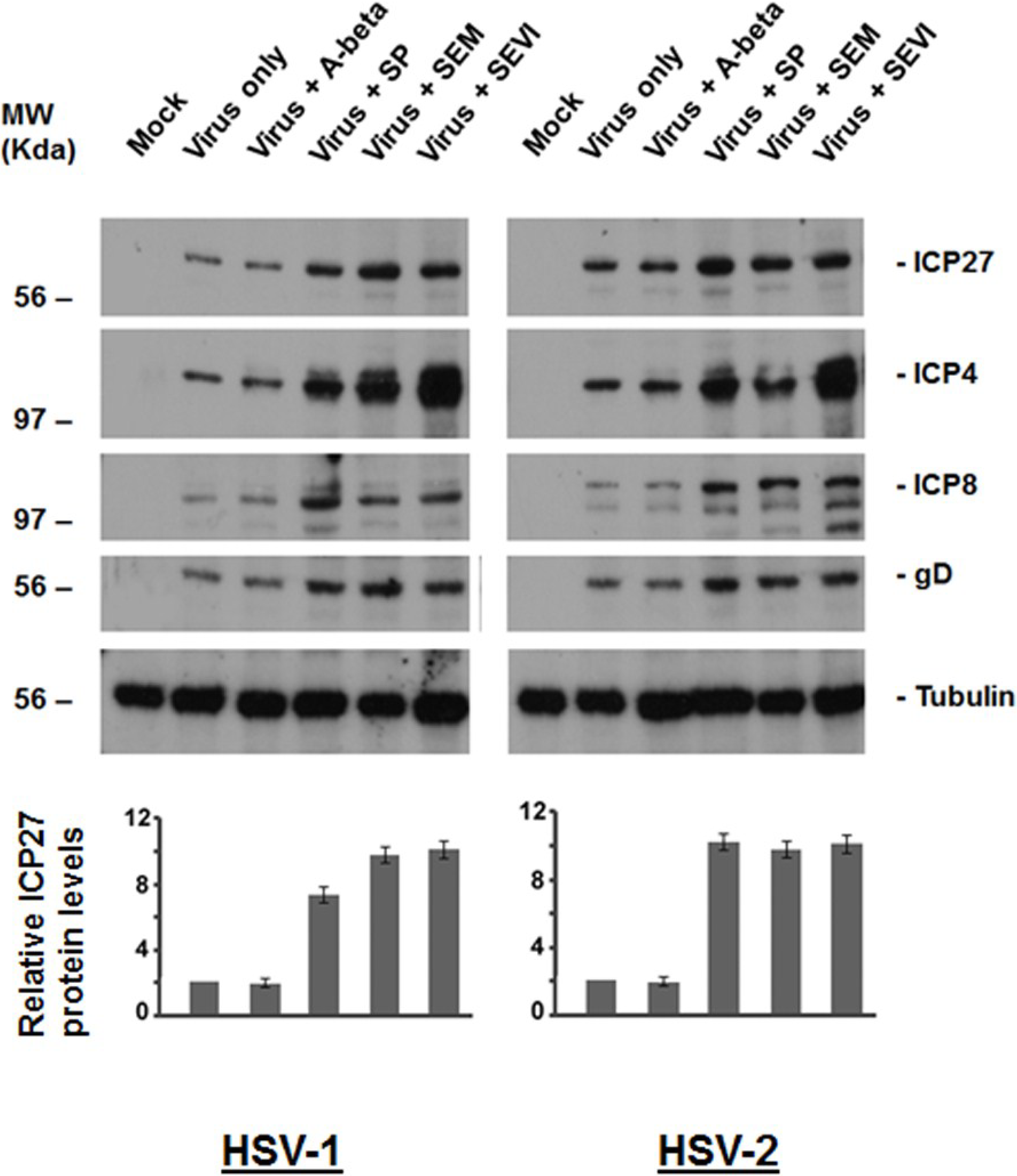
2.2. Viral Protein Production Was Increased by SEM and SEVI Amyloids and by SP


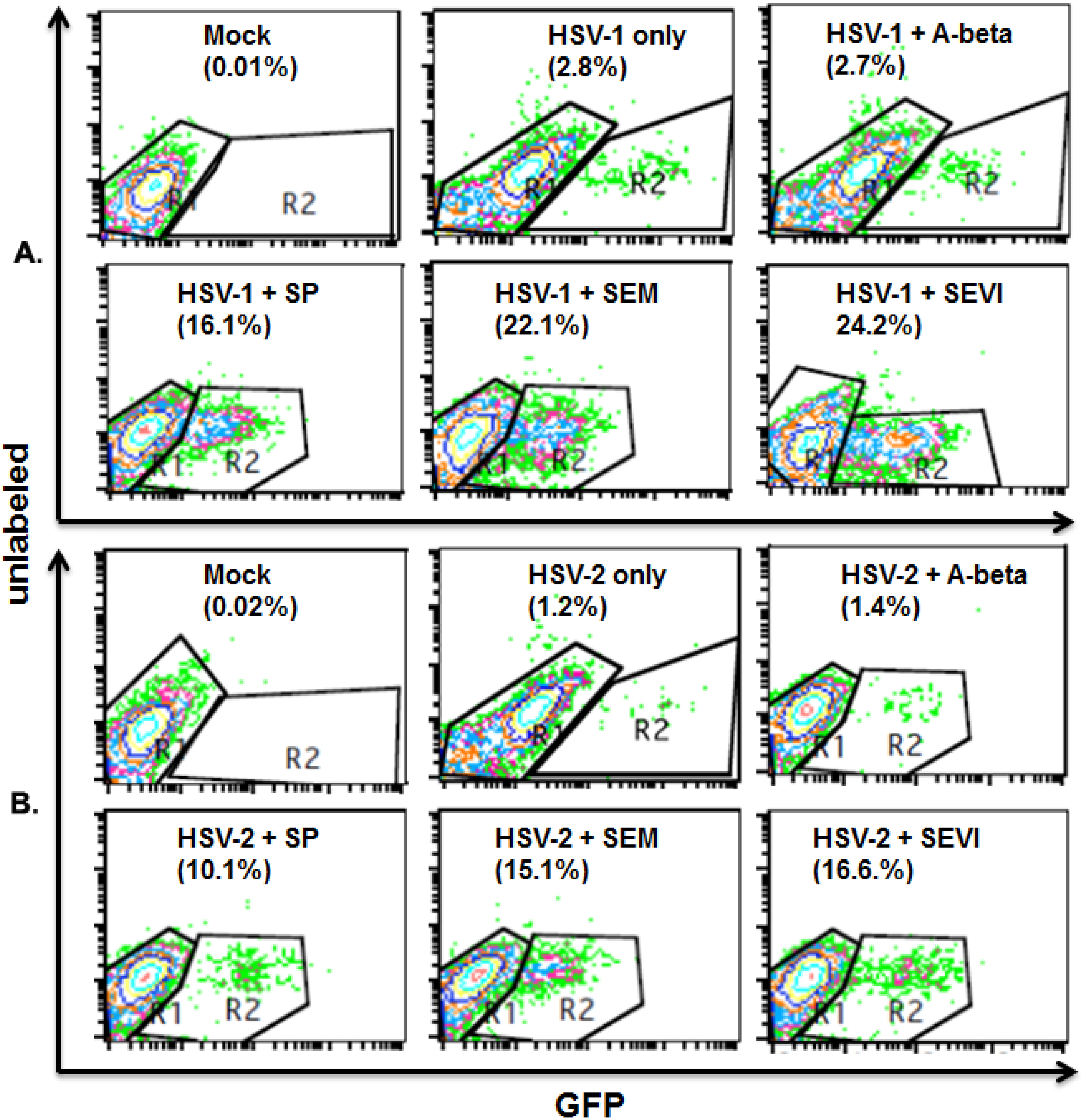
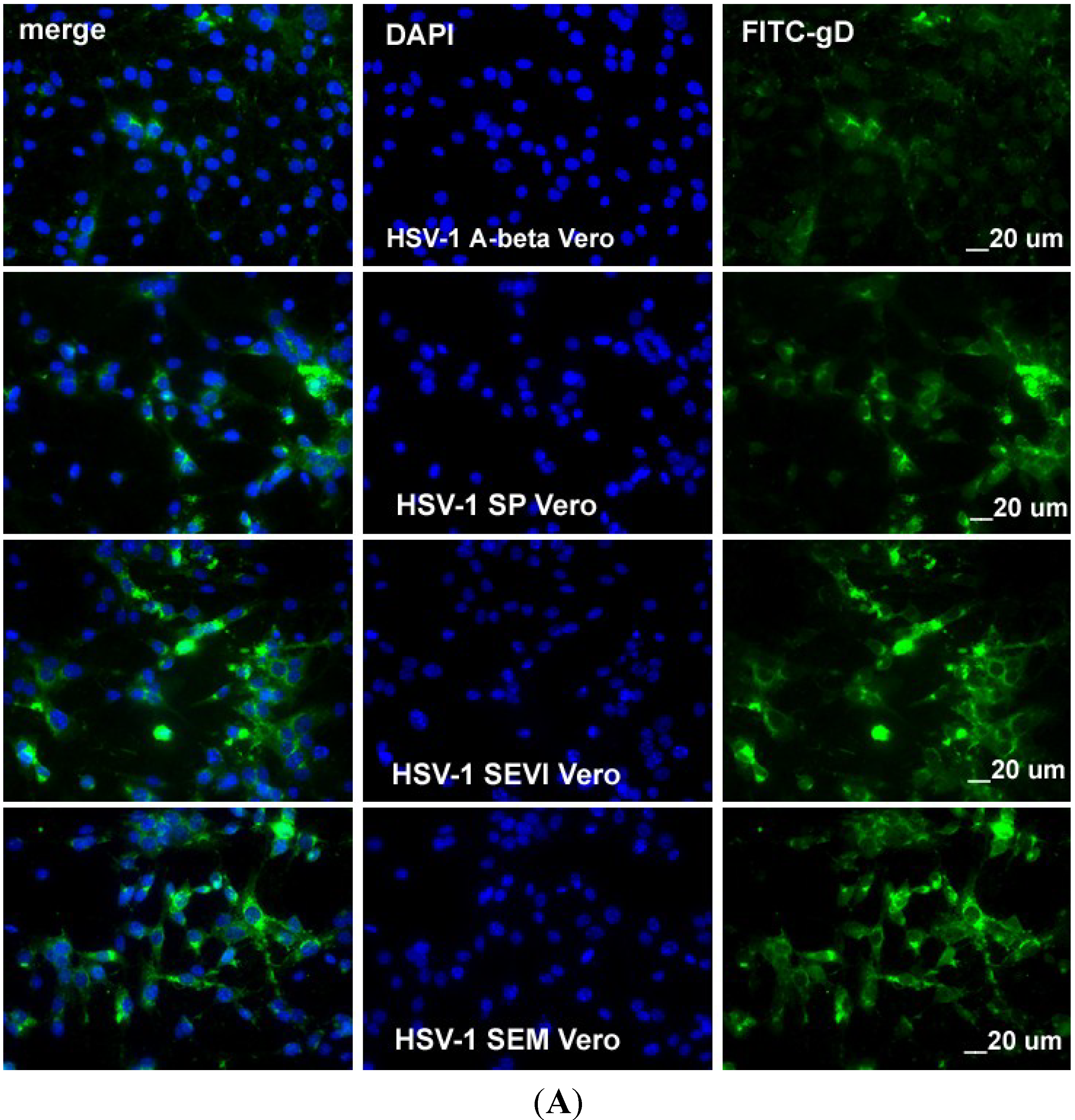
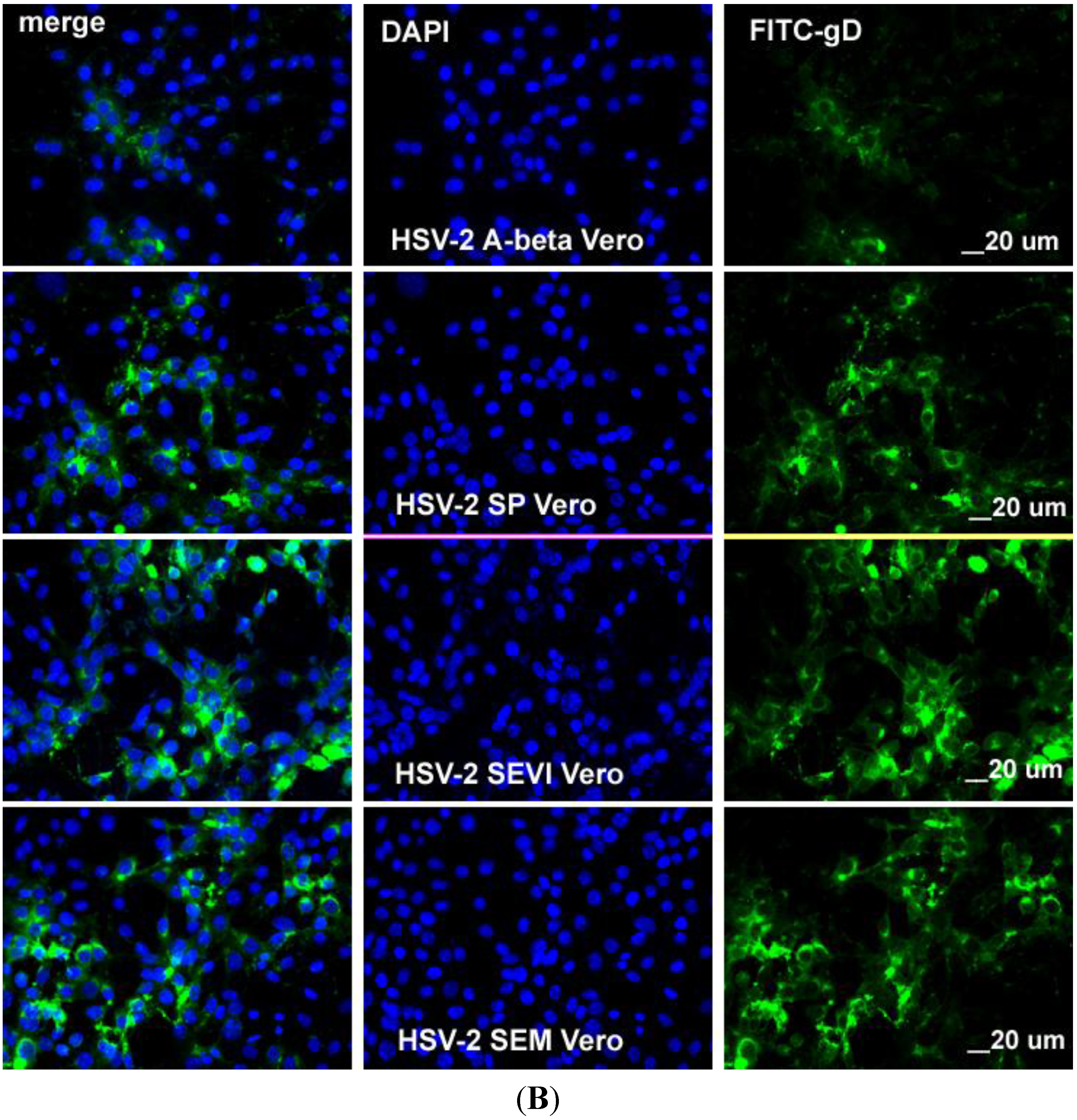
2.3. SEVI and SEM Amyloids Increase HSV Infection Rates
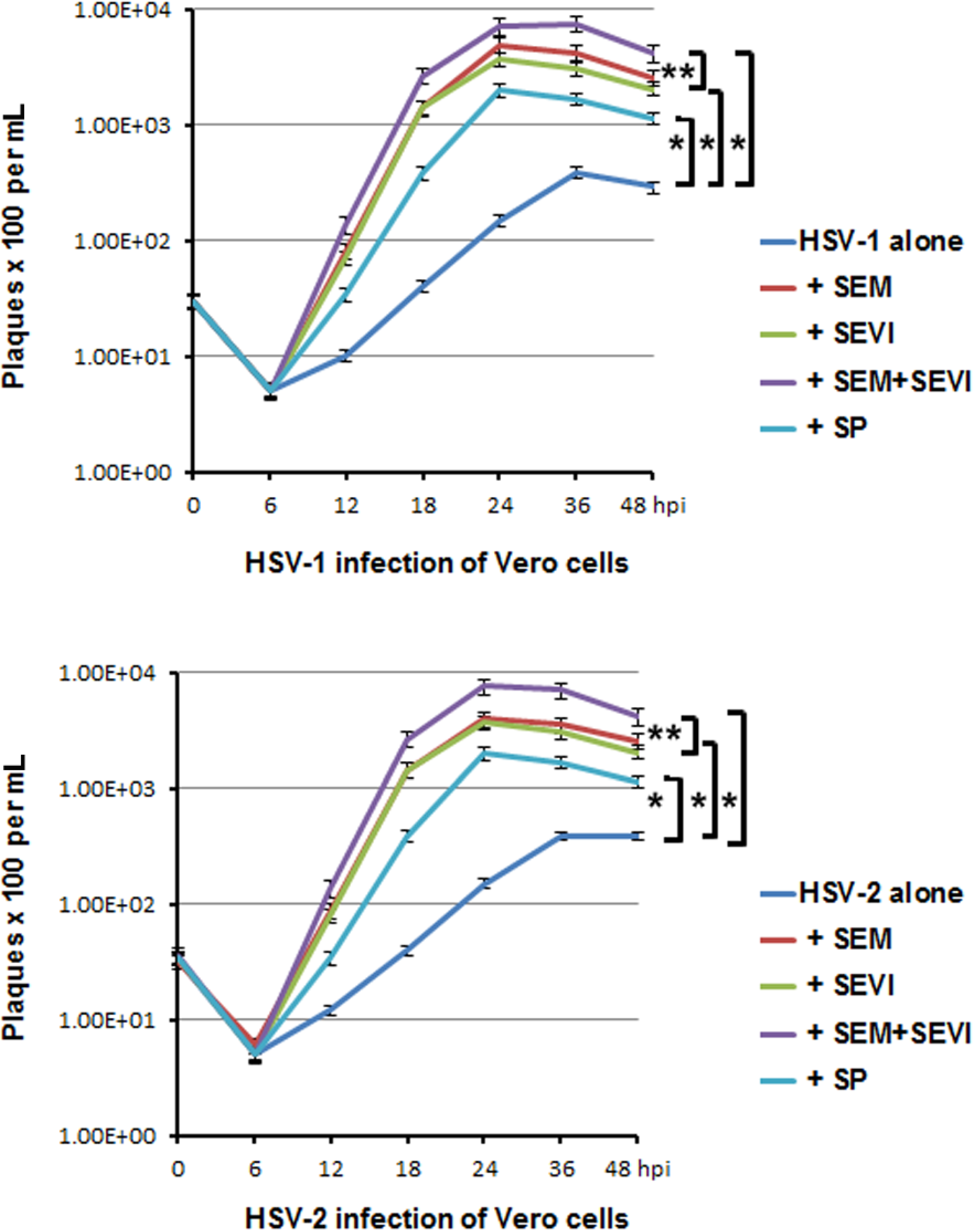
2.4. Viral Replication was Accelerated and Enhanced by SEM and SEVI Amyloids and by SP

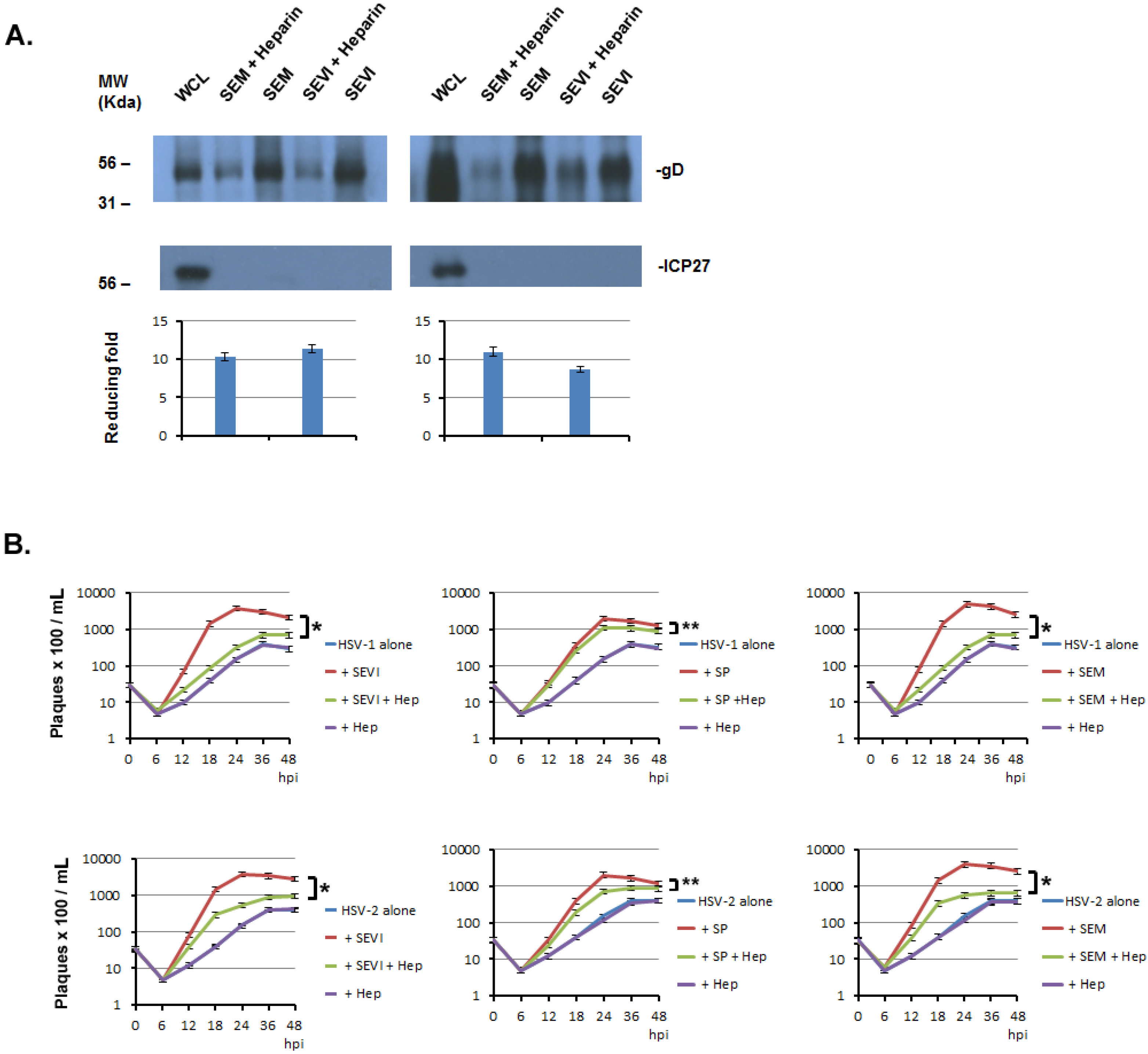
2.5. The Enhancement of SP, SEVI, and SEM in HSV Infections can Be Significantly Blocked by Heparin




3. Discussion
4. Materials and Methods
4.1. Tissue Culture and Viruses
4.2. Reagents
4.3. Antibodies
4.4. Purification of Viruses
4.5. Amyloid Fibril-Virus Binding Assay
4.6. Immunoblot Analysis
4.7. Cell visualization by Confocal Microscopy
4.8. Flow Cytometry
4.9. Plaque Formation Unit (pfu) Assay
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ochsendorf, F.R. Sexually transmitted infections: Impact on male fertility. Andrologia 2008, 40, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Muvunyi, C.M.; Masaisa, F.; Bayingana, C.; Mutesa, L.; Musemakweri, A.; Muhirwa, G.; Claeys, G.W. Decreased susceptibility to commonly used antimicrobial agents in bacterial pathogens isolated from urinary tract infections in Rwanda: Need for new antimicrobial guidelines. Am. J. Trop. Med. Hyg. 2011, 84, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Garolla, A.; Pizzol, D.; Bertoldo, A.; Menegazzo, M.; Barzon, L.; Foresta, C. Sperm viral infection and male infertility: Focus on HBV, HCV, HIV, HPV, HSV, HCMV, and AAV. J. Reprod. Immunol. 2013, 100, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Gimenes, F.; Souza, R.P.; Bento, J.C.; Teixeira, J.J.; Maria-Engler, S.S.; Bonini, M.G.; Consolaro, M.E. Male infertility: A public health issue caused by sexually transmitted pathogens. Nat. Rev. Urol. 2014, 11, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Chentoufi, A.A.; Benmohamed, L. Mucosal herpes immunity and immunopathology to ocular and genital herpes simplex virus infections. Clin. Dev. Immunol. 2012, 2012, e149135. [Google Scholar]
- Shin, H.; Iwasaki, A. Generating protective immunity against genital herpes. Trends Immunol. 2013, 34, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Hadigal, S.; Shukla, D. Exploiting herpes simplex virus entry for novel therapeutics. Viruses 2013, 5, 1447–1465. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Miranda-Saksena, M.; Douglas, M.W.; Cunningham, A.L. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 2008, 18, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Garland, S.M.; Tabrizi, S.N. Diagnosis of sexually transmitted infections (STI) using self-collected non-invasive specimens. Sex Health 2004, 1, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Looker, K.J.; Garnett, G.P.; Schmid, G.P. An estimate of the global prevalence and incidence of herpes simplex virus type 2 infection. Bull World Health Organ. 2008, 86, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.; Saracino, M.; Kuntz, S.; Magaret, A.; Selke, S.; Huang, M.L.; Schiffer, J.T.; Koelle, D.M.; Corey, L.; Wald, A. Standard-dose and high-dose daily antiviral therapy for short episodes of genital HSV-2 reactivation: Three randomised, open-label, cross-over trials. Lancet 2012, 379, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Bernard Roizman, D.; Richard, M.K.; Whitley, J. The family Herpesviridae: A Brief Introduction, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Munch, J.; Rucker, E.; Standker, L.; Adermann, K.; Goffinet, C.; Schindler, M.; Wildum, S.; Chinnadurai, R.; Rajan, D.; Specht, A.; et al. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell 2007, 131, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Roan, N.R.; Muller, J.A.; Liu, H.; Chu, S.; Arnold, F.; Sturzel, C.M.; Walther, P.; Dong, M.; Witkowska, H.E.; Kirchhoff, F.; et al. Peptides released by physiological cleavage of semen coagulum proteins form amyloids that enhance HIV infection. Cell Host Microbe. 2011, 10, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Roan, N.R.; Yamamura, Y. Seminal Plasma and Semen Amyloids Enhance Cytomegalovirus Infection in Cell Culture. J. Virol. 2013, 87, 12583–12591. [Google Scholar] [CrossRef] [PubMed]
- Roan, N.R.; Munch, J.; Arhel, N.; Mothes, W.; Neidleman, J.; Kobayashi, A.; Smith-McCune, K.; Kirchhoff, F.; Greene, W.C. The cationic properties of SEVI underlie its ability to enhance human immunodeficiency virus infection. J. Virol. 2009, 83, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Kaspersen, M.D.; Hollsberg, P. Seminal shedding of human herpesviruses. J. Virol. 2013, 10, 226. [Google Scholar] [CrossRef]
- Kapranos, N.; Petrakou, E.; Anastasiadou, C.; Kotronias, D. Detection of herpes simplex virus, cytomegalovirus, and Epstein-Barr virus in the semen of men attending an infertility clinic. Fertil. Steril. 2003, 79, 1566–1570. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Zeh, J.; Selke, S.; Warren, T.; Ryncarz, A.J.; Ashley, R.; Krieger, J.N.; Corey, L. Reactivation of genital herpes simplex virus type 2 infection in asymptomatic seropositive persons. N. Engl. J. Med. 2000, 342, 844–850. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding "one-dimensional crystallization" of amyloid: A pathogenic mechanism in Alzheimer's disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Blake, C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Adv. Protein. Chem. 1997, 50, 123–159. [Google Scholar] [PubMed]
- Uyangaa, E.; Patil, A.M.; Eo, S.K. Prophylactic and therapeutic modulation of innate and adaptive immunity against mucosal infection of herpes simplex virus. Immune Netw. 2014, 14, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Pirrone, V.; Wigdahl, B.; Krebs, F.C. The rise and fall of polyanionic inhibitors of the human immunodeficiency virus type 1. Antiviral. Res. 2011, 90, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Li, L.; Ishov, A.M.; Revol, V.; Epstein, A.L.; Maul, G.G. Determination of minimum herpes simplex virus type 1 components necessary to localize transcriptionally active DNA to ND10. J. Virol. 2003, 77, 5821–5828. [Google Scholar] [CrossRef] [PubMed]
- Yolamanova, M.; Meier, C.; Shaytan, A.K.; Vas, V.; Bertoncini, C.W.; Arnold, F.; Zirafi, O.; Usmani, S.M.; Muller, J.A.; Sauter, D.; et al. Peptide nanofibrils boost retroviral gene transfer and provide a rapid means for concentrating viruses. Nat. Nanotechnol. 2013, 8, 130–136. [Google Scholar] [CrossRef]
- Martinez, F.P.; Cosme, R.S.; Tang, Q. Murine cytomegalovirus major immediate-early protein 3 interacts with cellular and viral proteins in viral DNA replication compartments and is important for early gene activation. J. Gen. Virol. 2010, 91, 2664–2676. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, L.; Ortiz, T.; Tang, Q. Enhancement of Herpes Simplex Virus (HSV) Infection by Seminal Plasma and Semen Amyloids Implicates a New Target for the Prevention of HSV Infection. Viruses 2015, 7, 2057-2073. https://doi.org/10.3390/v7042057
Torres L, Ortiz T, Tang Q. Enhancement of Herpes Simplex Virus (HSV) Infection by Seminal Plasma and Semen Amyloids Implicates a New Target for the Prevention of HSV Infection. Viruses. 2015; 7(4):2057-2073. https://doi.org/10.3390/v7042057
Chicago/Turabian StyleTorres, Lilith, Tatiana Ortiz, and Qiyi Tang. 2015. "Enhancement of Herpes Simplex Virus (HSV) Infection by Seminal Plasma and Semen Amyloids Implicates a New Target for the Prevention of HSV Infection" Viruses 7, no. 4: 2057-2073. https://doi.org/10.3390/v7042057
APA StyleTorres, L., Ortiz, T., & Tang, Q. (2015). Enhancement of Herpes Simplex Virus (HSV) Infection by Seminal Plasma and Semen Amyloids Implicates a New Target for the Prevention of HSV Infection. Viruses, 7(4), 2057-2073. https://doi.org/10.3390/v7042057