
Characterization of Novel Transcripts in Pseudorabies Virus


Abstract
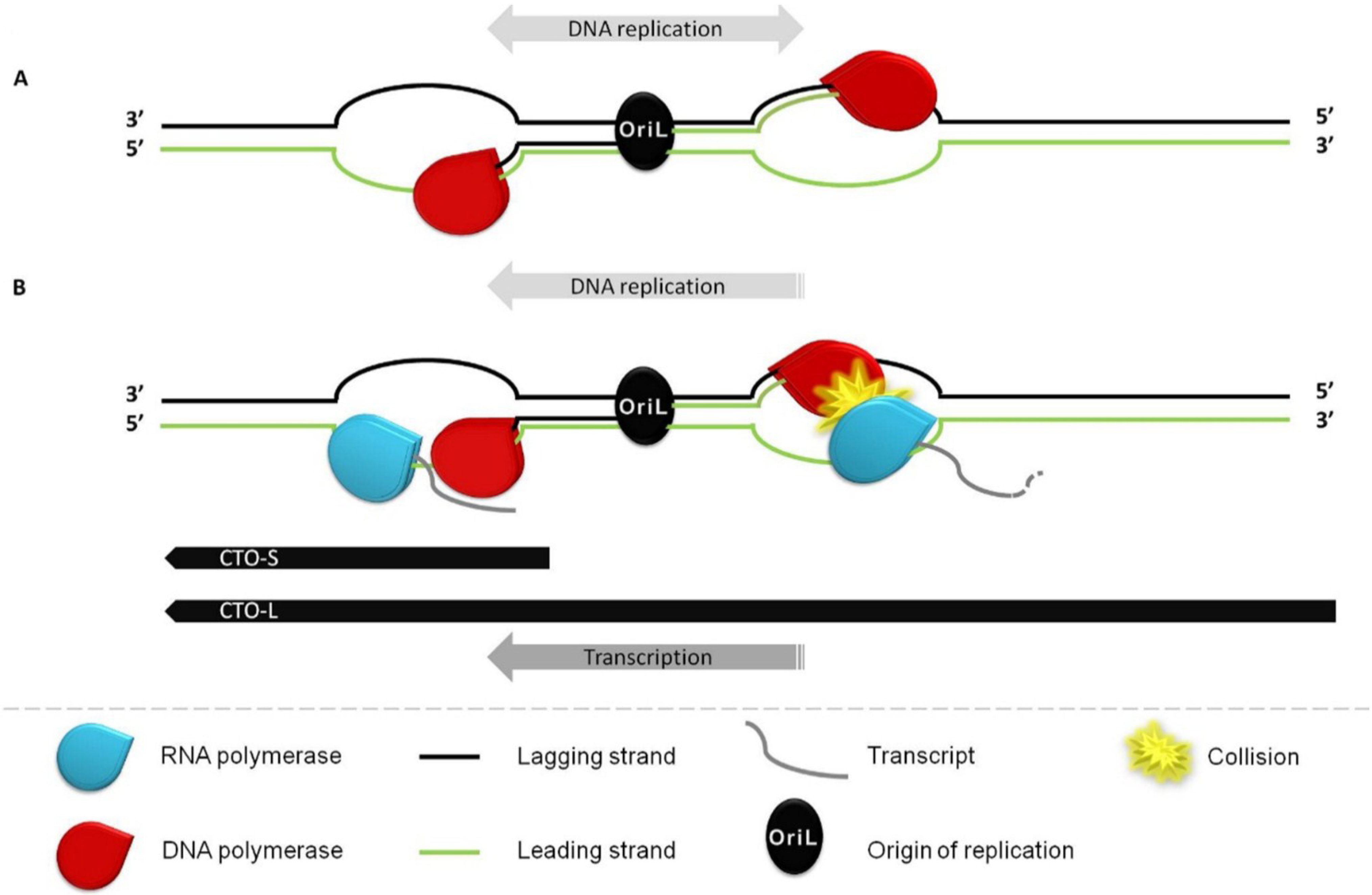
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Generation of Recombinant Viruses
2.3. RNA Isolation for RNA-Seq and Real-Time RT-PCR
2.4. Illumina HiScanSQ cDNA Sequencing
2.5. PacBio RS II cDNA Sequencing
2.5.1. PolyA RNA Purification
2.5.2. cDNA Synthesis
2.5.3. Library Preparation, Sequencing and Data Collection
2.6. Normalization of PacBio Data with Mitochondrial Transcripts
2.7. Reverse Transcription
2.8. Real-Time PCR

{kind=link}
{kind=link}
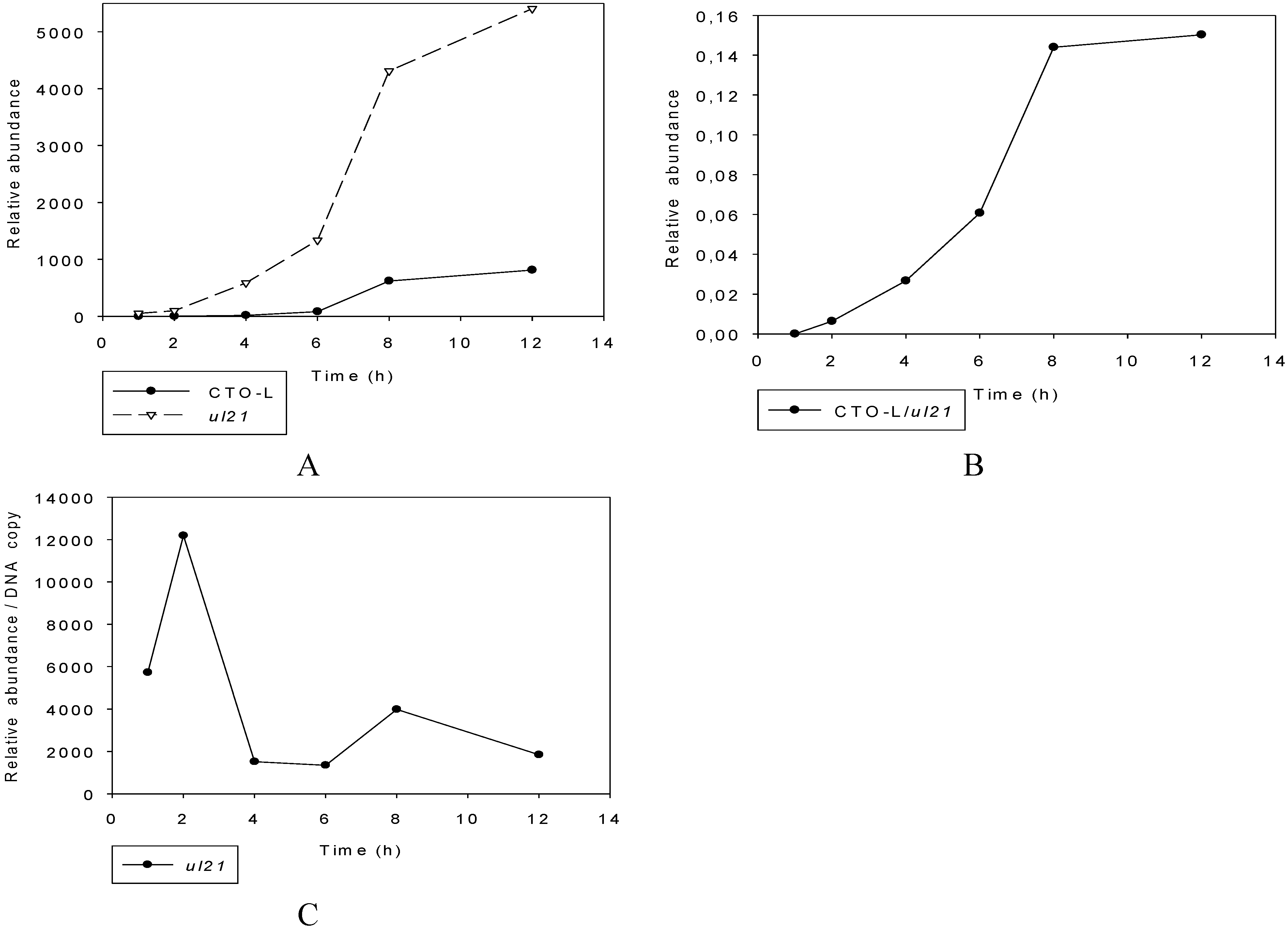{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′-3′) | Genomic Position | |
|---|---|---|---|
| A | CTO-S fw | GACGATCCGGCGGTCCCA | 63858–63875 |
| CTO-S rev | GCGCCACAACCCGGAGC | 63915–63931 | |
| CTO-L fw | GTG TCG CGG ACA GAG AAT GG | 64604–64623 | |
| CTO-L rev | GGC CCA GTA CCT GTT TCA GC | 64708–64727 | |
| B | T7-CTO-out fw | TAATACGACTCACTATAGGGAGAGGTCTCTAAGGGGGAACCAG | 63605–63626 |
| SP6-CTO-out rev | ATTTAGGTGACACTATAGAAGNGCCGAAAAATTCGCACATACC | 63989–64008 |
2.9. Treatment of Cells with CHX
2.10. Northern Blot Analysis
3. Results
3.1. Identification and Structural Characterization of Novel lncRNAs in PRV

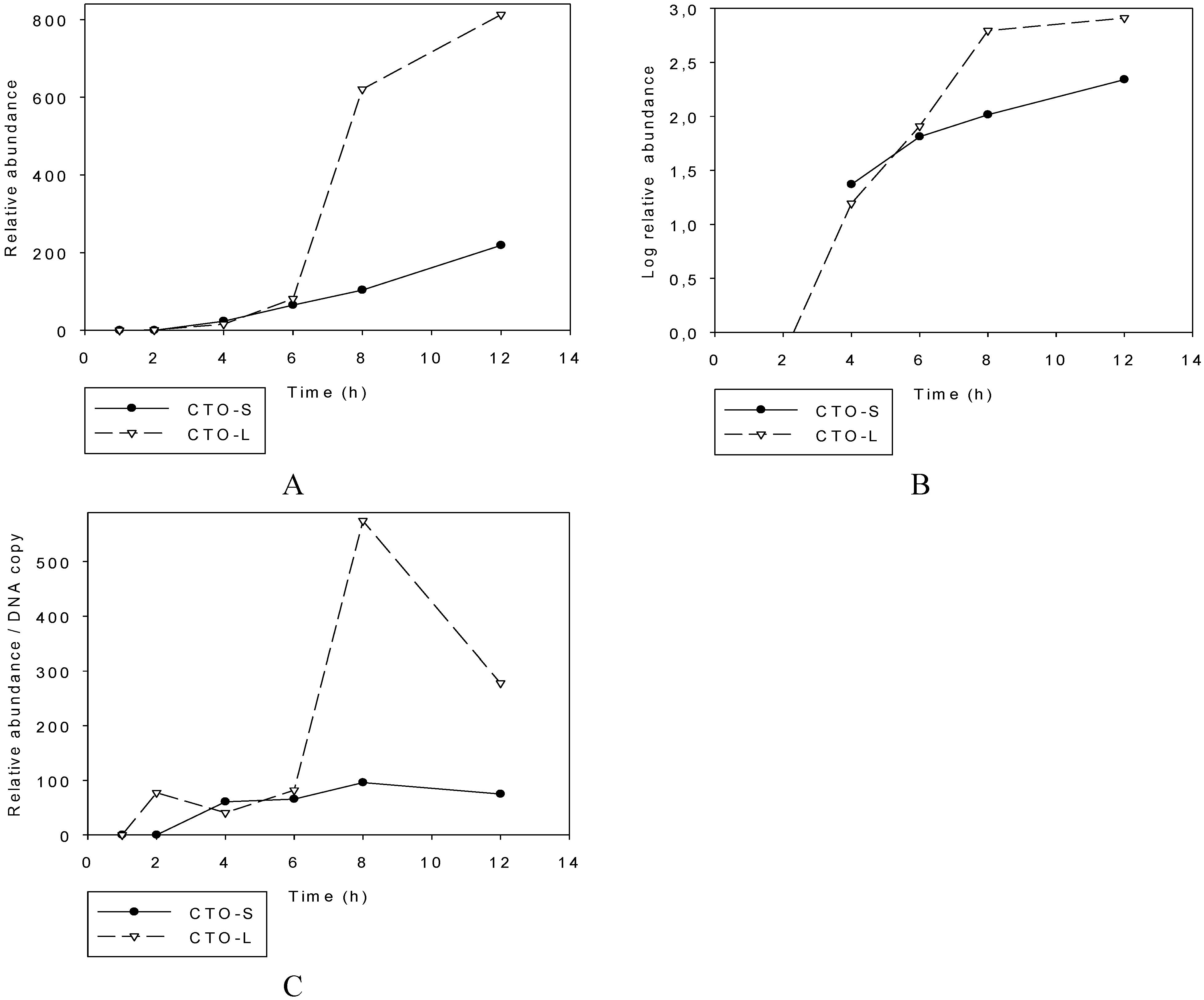
3.2. Transcriptional Analysis of CTO
3.2.1. Illumina RNA-Seq Analysis
3.2.2. PacBio RNA-Seq Analysis


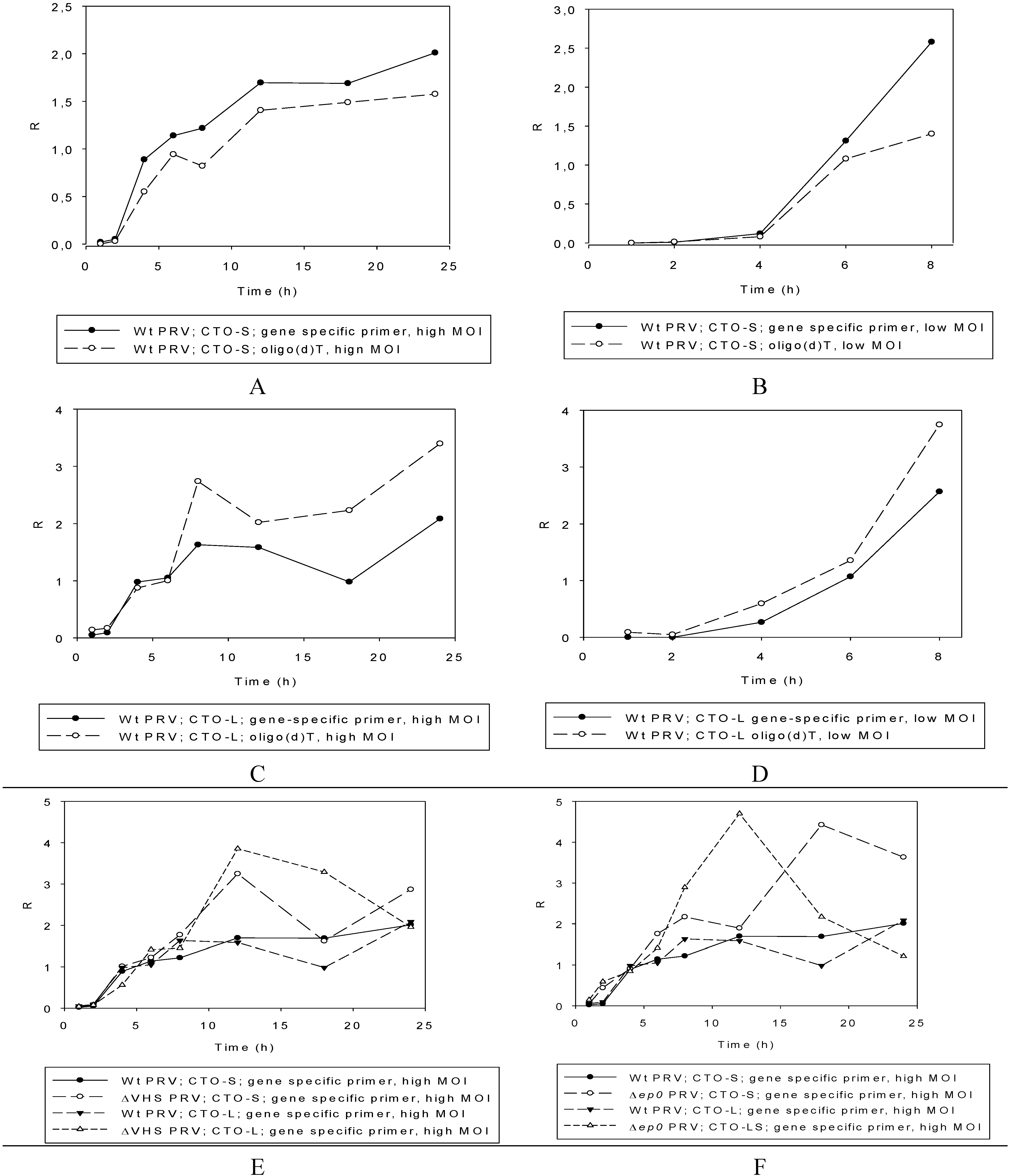
3.2.3. Multi-Time-Point Real-Time RT-PCR Analysis of CTOs in Wild-Type (wt) and Mutant Backgrounds
Wild-type PRV

Mutant PRVs
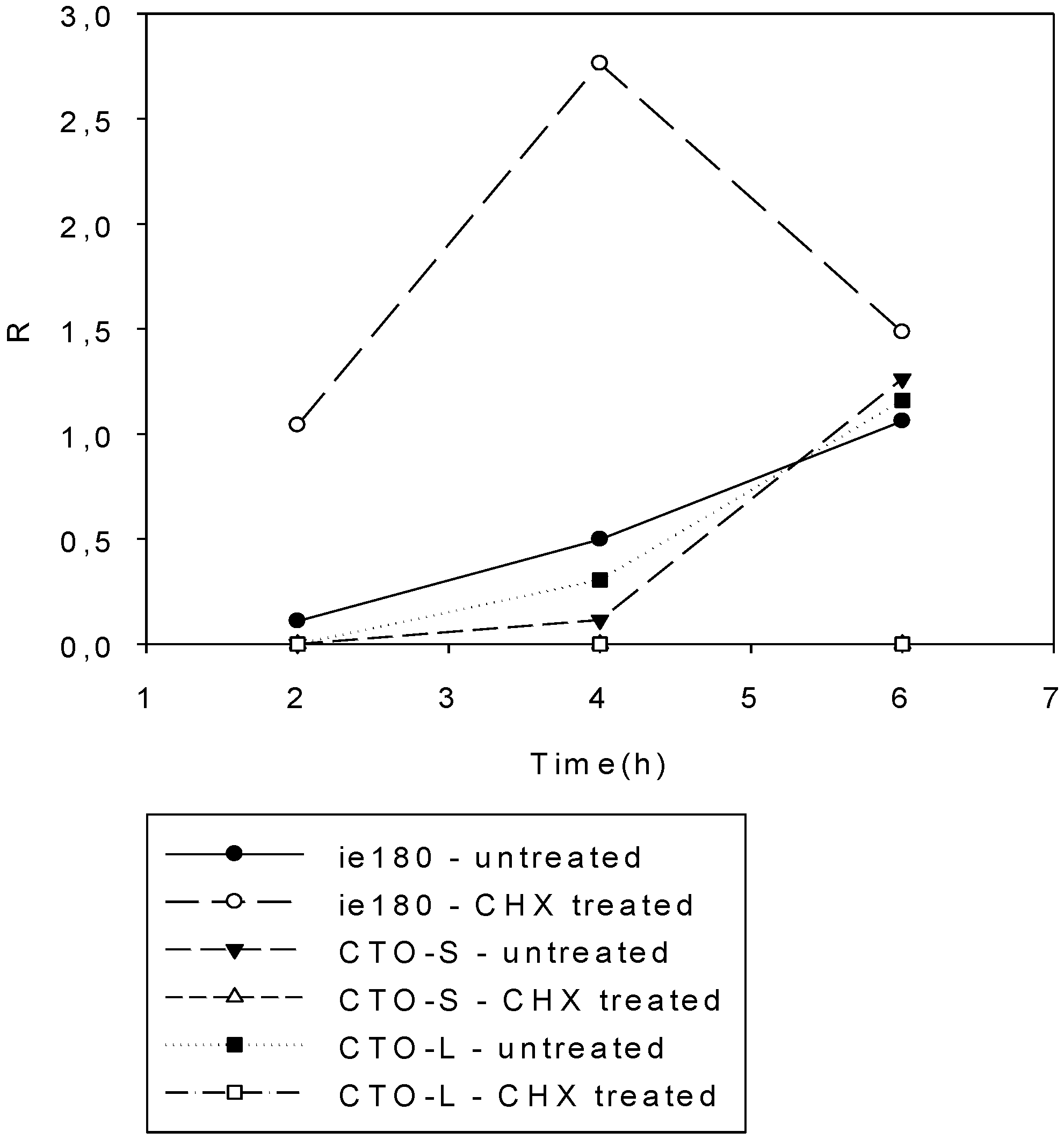
3.3. CTO Expression is Controlled by the IE180 Transactivator of PRV

3.4. CTO Expression in the Presence of an Inhibitor of DNA Replication
3.5. Northern Blot and in Silico Analyses Revealed that CTO-S is not a miRNA Precursor
4. Discussion

Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef] [PubMed]
- Szpara, M.L.; Kobiler, O.; Enquist, L.W. A common neuronal response to alphaherpesvirus infection. J. Neuroimmune Pharmacol. 2010, 5, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Strack, A.M. Pseudorabies virus as a transneuronal tract tracing tool: Specificity and applications to the sympathetic nervous system. Gene Ther. 1994, 1, S11–S14. [Google Scholar] [PubMed]
- Card, J.P.; Enquist, L.W. Transneuronal circuit analysis with pseudorabies viruses. Curr. Protoc. Neurosci. 2001, 1. [Google Scholar] [CrossRef]
- Boldogkői, Z.; Sík, A.; Dénes, A.; Reichart, A.; Toldi, J.; Gerendai, I.; Kovács, K.J.; Palkovits, M. Novel tracing paradigms—Genetically engineered herpesviruses as tools for mapping functional circuits within the CNS: Present status and future prospects. Prog. Neurobiol. 2004, 72, 417–445. [Google Scholar] [CrossRef] [PubMed]
- Boldogkői, Z.; Bálint, K.; Awatramani, G.B.; Balya, D.; Busskamp, V.; Viney, T.J.; Lagali, P.S.; Duebel, J.; Pásti, E.; Tombácz, D.; et al. Genetically timed, Activity sensor and Rainbow transsynaptic viral tools. Nat. Methods 2009, 6, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Prorok, J.; Kovács, P.P.; Kristóf, A.A.; Nagy, N.; Tombácz, D.; Tóth, J.S.; Ördög, B.; Jost, N.; Virág, L.; Papp, J.G.; et al. Herpesvirus-mediated delivery of a genetically encoded fluorescent Ca2+ sensor to canine cardiomyocytes. J. Biomed. Biotechnol. 2009, 2009. [Google Scholar] [CrossRef]
- Janowski, B.A.; Kaihatsu, K.; Huffman, K.E.; Schwartz, J.C.; Ram, R.; Hardy, D.; Mendelson, C.R.; Corey, D.R. Inhibiting transcription of chromosomal DNA with antigene peptide nucleic acids. Nat. Chem. Biol. 2005, 1, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [PubMed]
- Anselmo, A.; Flori, L.; Jaffrezic, F.; Rutigliano, T.; Cecere, M.; Cortes-Perez, N.; Lefèvre, F.; Rogel-Gaillard, C.; Giuffra, E. Co-expression of host and viral microRNAs in porcine dendritic cells infected by the pseudorabies virus. PLoS ONE 2011, 6, e17374. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Q.; Chen, D.J.; He, H.B.; Chen, D.S.; Chen, L.L.; Chen, H.C.; Liu, Z.F. Pseudorabies virus infected porcine epithelial cell line generates a diverse set of host microRNAs and a special cluster of viral microRNAs. PLoS ONE 2012, 7, e30988. [Google Scholar] [CrossRef] [PubMed]
- Grey, F.; Antoniewicz, A.; Allen, E.; Saugstad, J.; McShea, A.; Carrington, J.C.; Nelson, J. Identification and characterization of human cytomegalovirus-encoded microRNAs. J. Virol. 2005, 79, 12095–12099. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Zavolan, M.; Grässer, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; Tuschl, T. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Ramchandran, R. Natural antisense transcript: A concomitant engagement with protein-coding transcript. Oncotarget 2010, 1, 447–452. [Google Scholar] [PubMed]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell. 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W.; et al. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Miller, G. Polyadenylylated nuclear RNA encoded by Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1996, 93, 11883–11888. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Tarrant-Elorza, M.; Verma, S.; Purushothaman, P.; Pari, G.S. Regulation of viral and cellular gene expression by Kaposi’s sarcoma associated herpesvirus polyadenylated nuclear RNA. J. Virol. 2013, 87, 5540–5553. [Google Scholar] [CrossRef] [PubMed]
- Stroop, W.G.; Rock, D.L.; Fraser, N.W. Localization of herpes simplex virus in the trigeminal and olfactory systems of the mouse central nervous system during acute and latent infections by in situ hybridization. Lab. Investig. 1984, 51, 27–38. [Google Scholar] [PubMed]
- Cheung, A.K. Detection of pseudorabies virus transcripts in trigeminal ganglia of latently infected swine. J. Virol. 1989, 63, 2908–2913. [Google Scholar] [PubMed]
- Ward, P.L.; Barker, D.E.; Roizman, B. A novel herpes simplex virus 1 gene, UL43.5, maps antisense to the UL43 gene and encodes a protein which colocalizes in nuclear structures with capsid proteins. J. Virol. 1996, 70, 2684–2690. [Google Scholar] [PubMed]
- Chang, Y.E.; Menotti, L.; Filatov, F.; Campadelli-Fiume, G.; Roizman, B. UL27.5 is a novel γ2 gene antisense to the herpes simplex virus 1 gene encoding glycoprotein B. J. Virol. 1998, 72, 6056–6064. [Google Scholar] [PubMed]
- Jovasevic, V.; Roizman, B. The novel HSV-1 US5-1 RNA is transcribed off a domain encoding US5, US4, US3, US2 and α22. Virol. J. 2010, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Raghavan, B.; Kotur, M.; Cheatham, J.; Sedmak, D.; Cook, C.; Waldman, J.; Trgovcich, J. Antisense transcription in the human cytomegalovirus transcriptome. J. Virol. 2007, 81, 11267–11281. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D.; Takada, K. Role of EBERs in the pathogenesis of EBV infection. Adv. Cancer Res. 2010, 107, 119–136. [Google Scholar] [PubMed]
- Tombácz, D.; Tóth, J.S.; Boldogkői, Z. Deletion of the virion host shut: Off gene of pseudorabies virus results in selective upregulation of the expression of early viral genes in the late stage of infection. Genomics 2011, 98, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, D.; Tóth, J.S.; Boldogkoi, Z. Effects of deletion of the early protein 0 gene of pseudorabies virus on the overall viral gene expression. Gene 2012, 493, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, K.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.D.; Frenkel, N. The herpes simplex virus virion host shutoff function. J. Virol. 1989, 63, 4834–4839. [Google Scholar] [PubMed]
- Lin, H.W.; Chang, Y.Y.; Wong, M.L.; Lin, J.W.; Chang, T.J. Functional analysis of virion host shutoff protein of pseudorabies virus. Virology 2004, 324, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Saffran, H.A.; Pare, J.M.; Corcoran, J.A.; Weller, S.K.; Smiley, J.R. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 2007, 8, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, D.; Tóth, J.S.; Petrovszki, P.; Boldogkői, Z. Whole-genome analysis of pseudorabies virus gene expression by real-time quantitative RT-PCR assay. BMC Genomics 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Roizman, B. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J. Virol. 2006, 80, 9341–9345. [Google Scholar] [CrossRef] [PubMed]
- Tempel, S.; Tahi, F. A fast ab-initio method for predicting miRNA precursors in genomes. Nucleic Acids Res. 2012, 40, e80. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Wu, H.; Wang, W.; Ma, W.; Sun, X.; Lu, Z. MiPred: Classification of real and pseudo microRNA precursors using random forest prediction model with combined features. Nucleic Acids Res. 2007, 35, W339–W344. [Google Scholar] [CrossRef] [PubMed]
- Huvet, M.; Nicolay, S.; Touchon, M.; Audit, B.; d’Aubenton Carafa, Y.; Arneodo, A.; Thermes, C. Human gene organization driven by the coordination of replication and transcription. Genome Res. 2007, 17, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Necsulea, A.; Guillet, C.; Cadoret, J.C.; Prioleau, M.N.; Duret, L. The relationship between DNA replication and human genome organization. Mol. Biol. Evol. 2009, 26, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.A.; Weller, S.K. HSV-1 DNA replication. In Alpharpesviruses; Caister Academic Press: Norfolk, UK, 2011; pp. 89–112. [Google Scholar]
- Gu, Z.; Dong, J.; Wang, J.; Hou, C.; Sun, H.; Yang, W.; Bai, J.; Jiang, P. A novel inactivated gE/gI deleted pseudorabies virus (PRV) vaccine completely protects pigs from an emerged variant PRV challenge. Virus Res. 2015, 195, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, N.; Cong, X.; Wang, C.H.; Du, M.; Li, L.; Zhao, B.; Yuan, J.; Liu, D.D.; Li, S.; et al. Pathogenicity and genomic characterization of a pseudorabies virus variant isolated from Bartha-K61-vaccinated swine population in China. Vet. Microbiol. 2014, 174, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Osato, N.; Suzuki, Y.; Ikeo, K.; Gojobori, T. Transcriptional interferences in cis natural antisense transcripts of humans and mice. Genetics 2007, 176, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Gullerova, M.; Proudfoot, N.J. Convergent transcription induces transcriptional gene silencing in fission yeast and mammalian cells. Nat. Struct. Mol. Biol. 2012, 19, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Boldogkői, Z. Transcriptional interference networks coordinate the expression of functionally related genes clustered in the same genomic loci. Front. Genet. 2012, 3. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tombácz, D.; Csabai, Z.; Oláh, P.; Havelda, Z.; Sharon, D.; Snyder, M.; Boldogkői, Z. Characterization of Novel Transcripts in Pseudorabies Virus. Viruses 2015, 7, 2727-2744. https://doi.org/10.3390/v7052727
Tombácz D, Csabai Z, Oláh P, Havelda Z, Sharon D, Snyder M, Boldogkői Z. Characterization of Novel Transcripts in Pseudorabies Virus. Viruses. 2015; 7(5):2727-2744. https://doi.org/10.3390/v7052727
Chicago/Turabian StyleTombácz, Dóra, Zsolt Csabai, Péter Oláh, Zoltán Havelda, Donald Sharon, Michael Snyder, and Zsolt Boldogkői. 2015. "Characterization of Novel Transcripts in Pseudorabies Virus" Viruses 7, no. 5: 2727-2744. https://doi.org/10.3390/v7052727
APA StyleTombácz, D., Csabai, Z., Oláh, P., Havelda, Z., Sharon, D., Snyder, M., & Boldogkői, Z. (2015). Characterization of Novel Transcripts in Pseudorabies Virus. Viruses, 7(5), 2727-2744. https://doi.org/10.3390/v7052727