
Hepatitis E Virus Genotype 3 Diversity: Phylogenetic Analysis and Presence of Subtype 3b in Wild Boar in Europe

Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and RNA Extraction
2.2. Primers and Probe Design
2.3. PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region Name and Internal Length (nt) | Primer Name | Position | Sequence | Step | Product Length (bp) |
|---|---|---|---|---|---|
| ORF1 -5´ This study | HEV.ORF1_F1 | 33–58 | CCCAYCAGTTYATWAAGGCTCCTGGC | RT-PCR | 493 |
| HEV.ORF1_R1 | 497–525 | TGCARDGARTANARRGCNAYNCCNGTCTC | |||
| HEV.ORF1_F2 | 98–126 | AAYTCYGCCYTGGCGAATGCTGTGGTGGT | nested PCR | 302 | |
| HEV.ORF1_R2 | 377–399 | CCVCGRGTNGGRGCRGWRTACCA | |||
| HVR (for genotype 3) This study | HEV.HVR_F1 | 2069–2091 | TTYTCYCCTGGGCAYMTYTGGGA | RT-PCR | 401 |
| HEV.HVR_R1 | 2441–2469 | TTAACCARCCARTCACARTCYGAYTCAAA | |||
| HEV.HVR_F2a | 2135–2157 | ACYTGGTCHACATCTGGYTTYTC | nested PCR | 263 or 293 | |
| HEV.HVR_F2b | 2165–2184 | TTYTCCCCYCCTGAGGCGGC | |||
| HEV.HVR_R2 | 2405–2427 | TACACCTTRGCSCCRTCRGGRTA | |||
| RdRp 280 (4312–4591) (Johne et al. 2010) | HEV-cs | 4181–4203 | TCGCGCATCACMTTYTTCCARAA | RT-PCR | 469 |
| HEV-cas | 4628–4650 | GCCATGTTCCAGACDGTRTTCCA | |||
| HEV-csn | 4287–4311 | GTGCTCTGTTTGGCCCNTGGTTYMG | nested PCR | 330 | |
| HEV-casn | 4592–4617 | CCAGGCTCACCRGARTGYTTCTTCCA | |||
| ORF3 225 (5205–5429) This study | HEV.ORF3_F1 | 5126–5145 | MGGKTRGAATGAATAACATG | RT-PCR | 326 or 362 |
| HEV.ORF3_R2 | 5430–5451 | GGCGCTGGGAYTGGTCRCGCCA | |||
| HEV.ORF3_R1 | 5467–5487 | CAGYTGGGGYAGRTCGACGRC | |||
| HEV.ORF3_F2 | 5182–5204 | GGGCTGTTCTGTTKYTGYTCYTC | nested PCR | 219 or 269 | |
| HEV.ORF3_R2 | 5430–5451 | GGCGCTGGGAYTGGTCRCGCCA | |||
| HEV.ORF3_R2a | 5382–5401 | CGAGGGCGAGCTCCAGCCCC | |||
| modified Diagnostic-qPCR - This study and (Schlosser et al. 2014) | HEV.Fa | 5278–5294 | GTGCCGGCGGTGGTTTC | RT-qPCR | 81 |
| HEV.Fb | 5278–5296 | GTGCCGGCGGTGGTTTCTG | |||
| HEV.R | 5340–5359 | GCGAAGGGGTTGGTTGGATG | |||
| HEV.P | 5300–5320 | FAM-TGACMGGGTTGATTCTCAGCC-BHQ1 | |||
| ORF2 187 (6277–6488) This study | HEV.ORF2_F1 | 6205–6223 | CDGCNACYCGBTTYATGAA | RT-PCR | 393 |
| HEV.ORF2_R1a | 6573–6598 | GTKAGRGARAGCCAWAGYACATCATT | |||
| HEV.ORF2_R1b | 6573–6598 | GTRAGNGADAGCCACARRACATCATT | |||
| HEV.ORF2_F2 | 6276–6301 | GCBYTHACNYTRTTYAAYCTTGCTGA | nested PCR | 241 | |
| HEV.ORF2_R2 | 6489–6517 | TGYTCRTGYTGRTTRTCRTARTCYTGDAT |
2.4. Sequencing, Phylogenetic Analysis and Classification
3. Results
3.1. HEV RNA Detection
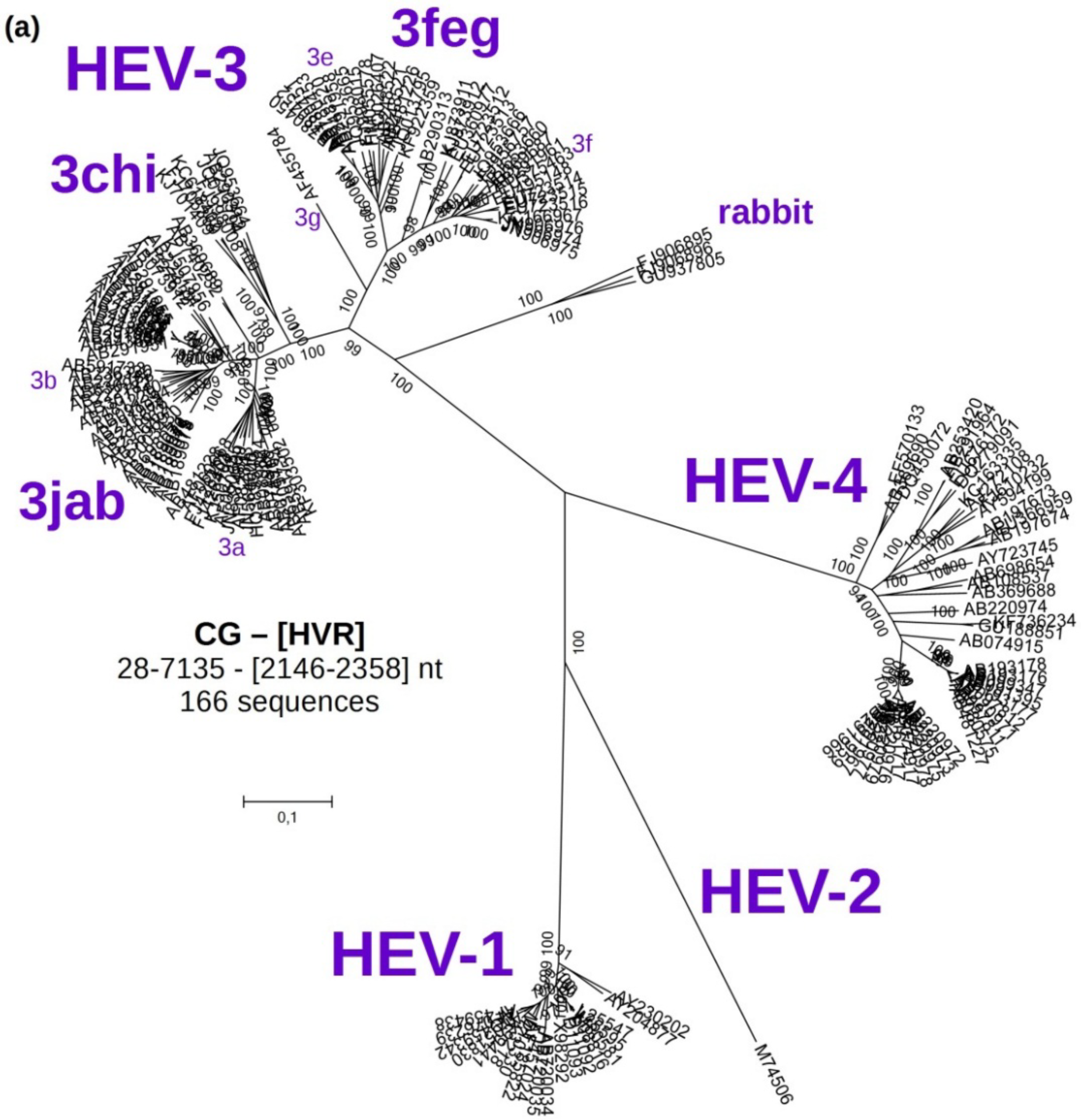
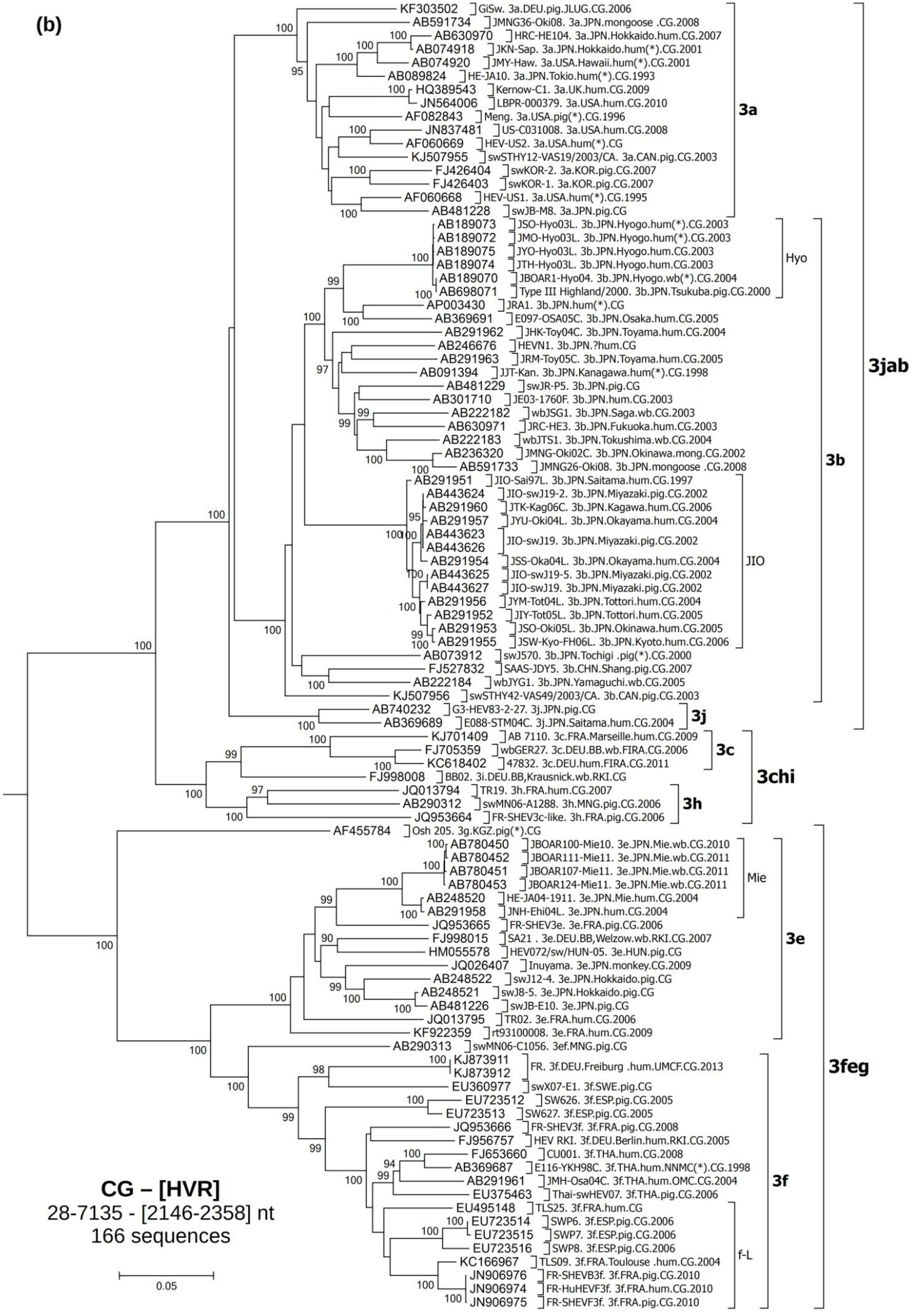
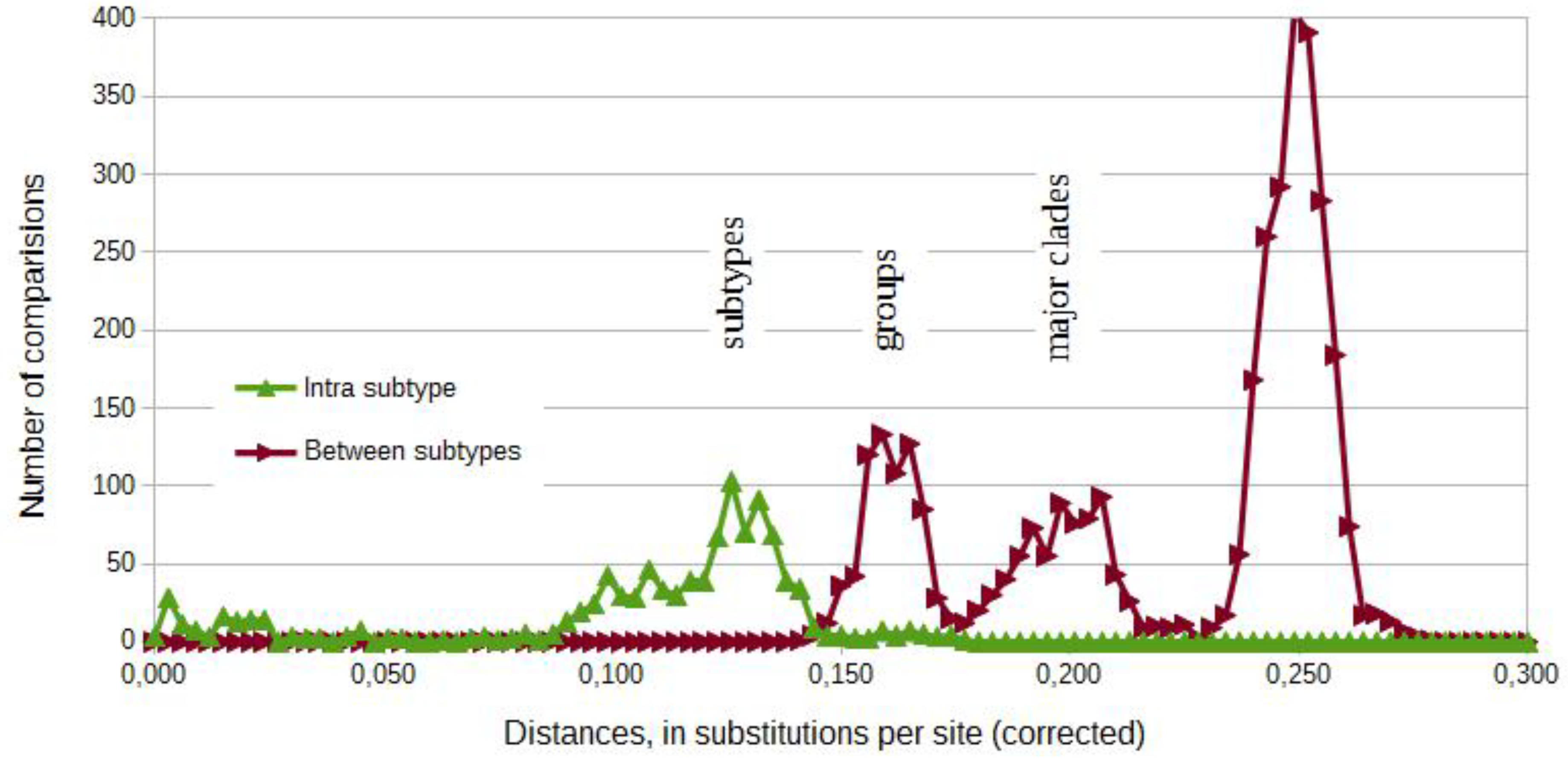
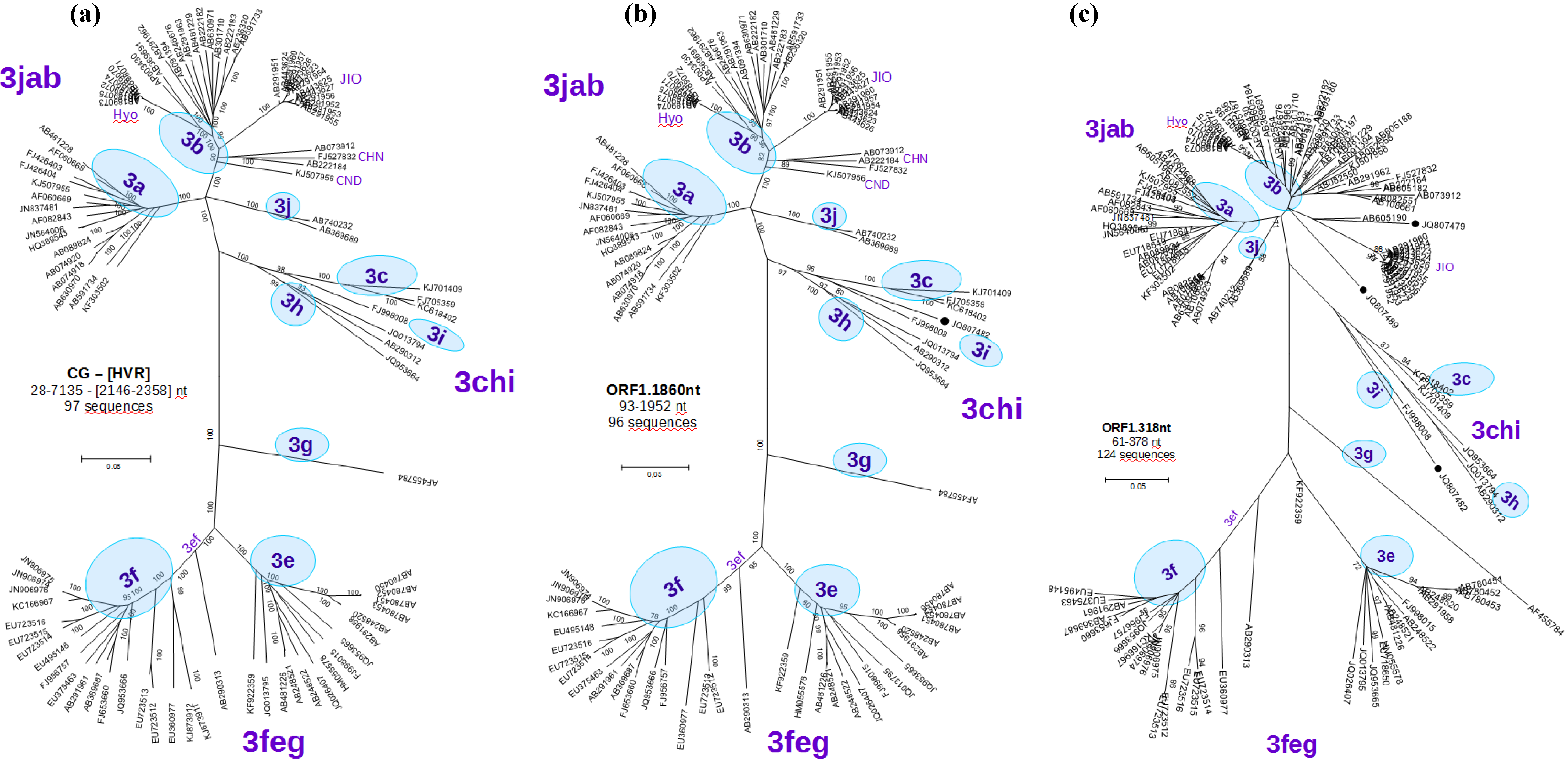
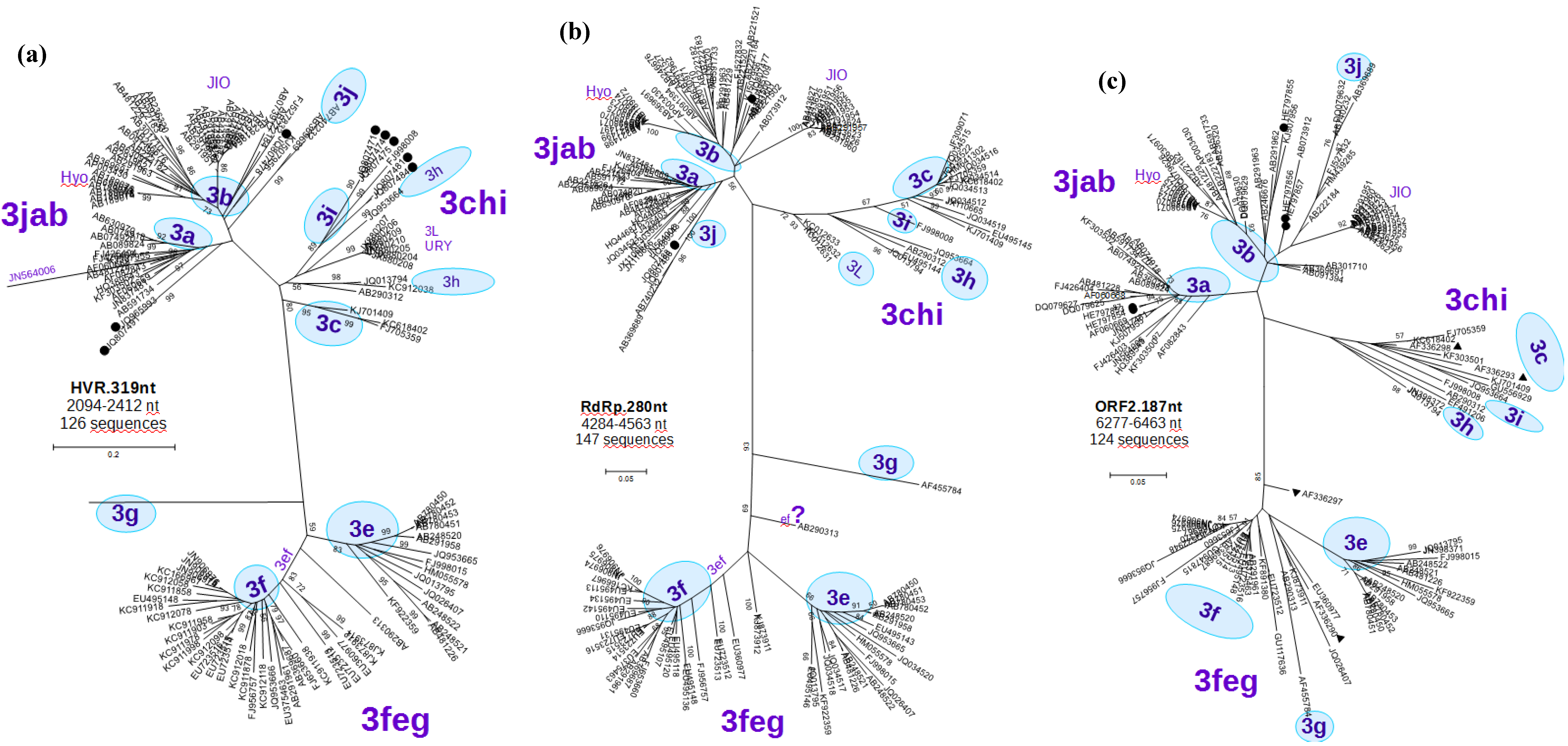
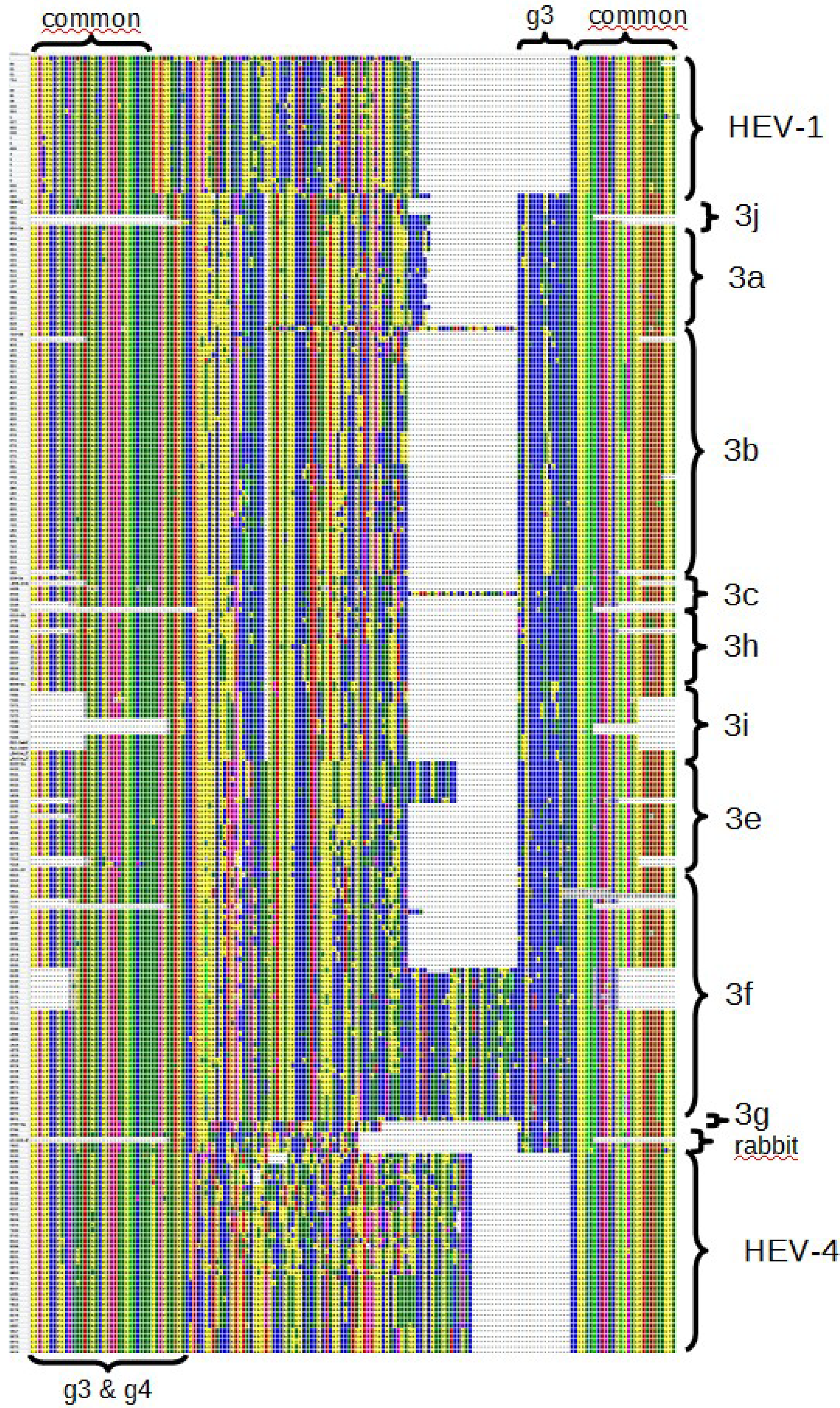
3.2. Phylogenetic Analyses






| Origen | 3 | 3jab | 3chi | 3feg | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 3b | 3d | 3j | 3h | 3L | 3i | 3c | 3e | 3ef | 3f | 3g | 3k | |||
| Africa | 7 | 7 | |||||||||||||
| CMR | 0’4 | 0’4 | |||||||||||||
| CAN | 0’3 | 0’3 | |||||||||||||
| America | 27 | 2 | 2 | 15 | 47 | ||||||||||
| ARG | 1 | 3’1 | 5’1 | ||||||||||||
| URY | 10 | 10 | |||||||||||||
| BRA | 0’1 | 0’1 | |||||||||||||
| CUB | 7’6 | 7’6 | |||||||||||||
| CAN | 2’1 | 1’1 | 3’2 | ||||||||||||
| MEX | 0’1 | 0’1 | |||||||||||||
| USA | 6’4’0’1 | 6’4’0’1 | |||||||||||||
| Asia | 18 | 67 | 3 | 3 | 3 | 13 | 1 | 5 | 1 | 114 | |||||
| CHN | 1’1 | 0’3 | 1’4 | ||||||||||||
| JPN | 10’3’2 | 30’12’23 | 1’2 | 4’3’4 | 46’20’29 | ||||||||||
| KOR | 0’2 | 0’2 | |||||||||||||
| KGZ | 0’1 | 0’1 | |||||||||||||
| MNG | 0’1 | 0’1 | 0’2 | ||||||||||||
| NZL | 1’1 | 1 | 2’1 | ||||||||||||
| THA | 4’1 | 4’1 | |||||||||||||
| Europe | 8 | 59 | 2 | 10 | 25 | 277 | 326 | 2 | 208 | 9 | 940 | ||||
| ESP | 0’0’0’1 | 0’0’0’1 | 0’2 | 7’27’0’13 | 7’29’0’15 | ||||||||||
| FRA | 1 | 3’1 | 3’1 | 4’1’0’1 | 24’9 | 35’12’0’1 | |||||||||
| GRC | 1 | 1 | 2’3 | ||||||||||||
| ITA | 1 | 0’1 | 0’2 | 1’3 | |||||||||||
| AUT | 0’4 | 1’1 | 1’5 | ||||||||||||
| NLD | 1’10 | 16’29’1 | 2’2’0’1 | 5’33’0’1 | 24’74’1’2 | ||||||||||
| DEU | 4 | 9’2’11 | 0’0’1 | 0’0’4 | 0’0’21 | 40’6’14 | 11’8’6 | 2 | 15’1’1 | 1 | 82’17’61 | ||||
| HUN | 0’8’3 | 3’12’2 | 2 | 5’20’5 | |||||||||||
| CZE | 2 | 1’4 | 1’4’2 | 4’8’2 | |||||||||||
| SRB | 0’4 | 0’1’2 | 0’5 | ||||||||||||
| SVN | 0’3 | 0’3 | |||||||||||||
| GBR | 3 | 6 | 165 | 268 | 57 | 507 | |||||||||
| SWE | 1 | 2 | 0’0’1 | 4’1 | 7’0’1’1 | ||||||||||
| Total | 8 | 104 | 71 | 3 | 5 | 20 | 15 | 25 | 277 | 339 | 3 | 213 | 10 | 3 | 1109 |
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Supplementary Information
Tables
Figures
Supplementary Files
Conflicts of Interest
References
- Moal, V.; Gerolami, R.; Ferretti, A.; Purgus, R.; Devichi, P.; Burtey, S.; Colson, P. Hepatitis e virus of subtype 3i in chronically infected kidney transplant recipients in southeastern france. J. Clin. Microbiol. 2014, 52, 3967–3972. [Google Scholar] [CrossRef] [PubMed]
- Moal, V.; Ferretti, A.; Devichi, P.; Colson, P. Genome sequence of a hepatitis e virus of genotype 3e from a chronically infected kidney transplant recipient. Genome Announc. 2014, 2, e01156–e0115613. [Google Scholar] [CrossRef] [PubMed]
- Marano, G.; Vaglio, S.; Pupella, S.; Facco, G.; Bianchi, M.; Calizzani, G.; Candura, F.; Catalano, L.; Farina, B.; Lanzoni, M.; et al. Hepatitis E: An old infection with new implications. Blood Transfus. = Trasfus. Sangue 2015, 13, 6–20. [Google Scholar]
- Colson, P.; Borentain, P.; Queyriaux, B.; Kaba, M.; Moal, V.; Gallian, P.; Heyries, L.; Raoult, D.; Gerolami, R. Pig liver sausage as a source of hepatitis e virus transmission to humans. J. Infect. Dis. 2010, 202, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Li, T.C.; Chijiwa, K.; Sera, N.; Ishibashi, T.; Etoh, Y.; Shinohara, Y.; Kurata, Y.; Ishida, M.; Sakamoto, S.; Takeda, N.; et al. Hepatitis e virus transmission from wild boar meat. Emerg. Infectious Dis. 2005, 11, 11–13. [Google Scholar] [CrossRef]
- Tei, S.; Kitajima, N.; Takahashi, K.; Mishiro, S. Zoonotic transmission of hepatitis e virus from deer to human beings. Lancet 2003, 362, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Renou, C.; Pariente, A.; Cadranel, J.F.; Nicand, E.; Pavio, N. Clinically silent forms may partly explain the rarity of acute cases of autochthonous genotype 3c hepatitis e infection in france. J. Clin. Virol. 2011, 51, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.C.; Wichmann, O.; Duizer, E. Transmission routes and risk factors for autochthonous hepatitis e virus infection in europe: A systematic review. Epidemiol. Infect. 2010, 138, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Meader, E.; Thomas, D.; Salmon, R.; Sillis, M. Seroprevalence of hepatitis e virus in the UK farming population. Zoonoses Public Health 2010, 57, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Kaba, M.; Davoust, B.; Marie, J.L.; Colson, P. Detection of hepatitis e virus in wild boar (sus scrofa) livers. Vet. J. 2010, 186, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Preiss, J.C.; Plentz, A.; Engelmann, E.; Schneider, T.; Jilg, W.; Zeitz, M.; Duchmann, R. Autochthonous hepatitis e virus infection in germany with sequence similarities to other european isolates. Infection 2006, 34, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Kaci, S.; Nöckler, K.; Johne, R. Detection of hepatitis e virus in archived german wild boar serum samples. Vet. Microbiol. 2008, 128, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Adlhoch, C.; Wolf, A.; Meisel, H.; Kaiser, M.; Ellerbrok, H.; Pauli, G. High hev presence in four different wild boar populations in east and west germany. Vet. Microbiol. 2009, 139, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Schielke, A.; Sachs, K.; Lierz, M.; Appel, B.; Jansen, A.; Johne, R. Detection of hepatitis e virus in wild boars of rural and urban regions in germany and whole genome characterization of an endemic strain. Virol. J. 2009, 6, e58. [Google Scholar] [CrossRef]
- Wenzel, J.J.; Preiß, J.; Schemmerer, M.; Huber, B.; Plentz, A.; Jilg, W. Detection of hepatitis e virus (hev) from porcine livers in southeastern germany and high sequence homology to human hev isolates. J. Clin. Virol. 2011, 52, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Fodor, D.; Forgách, P.; Kátai, A.; Szucs, G. Characterization and zoonotic potential of endemic hepatitis e virus (hev) strains in humans and animals in hungary. J. Clin. Virol. 2009, 44, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Ponterio, E.; di Bartolo, I.; Orru, G.; Liciardi, M.; Ostanello, F.; Ruggeri, F.M. Detection of serum antibodies to hepatitis e virus in domestic pigs in italy using a recombinant swine hev capsid protein. BMC Vet. Res. 2014, 10, e133. [Google Scholar] [CrossRef]
- Caruso, C.; Modesto, P.; Bertolini, S.; Peletto, S.; Acutis, P.L.; Dondo, A.; Robetto, S.; Mignone, W.; Orusa, R.; Ru, G.; et al. Serological and virological survey of hepatitis e virus in wild boar populations in northwestern italy: Detection of hev subtypes 3e and 3f. Arch. Virol. 2015, 160, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Van der Poel, W.H.; Verschoor, F.; van der Heide, R.; Herrera, M.I.; Vivo, A.; Kooreman, M.; de Roda Husman, A.M. Hepatitis e virus sequences in swine related to sequences in humans, the netherlands. Emerg. Infect. Dis. 2001, 7, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Rutjes, S.A.; Lodder, W.J.; Lodder-Verschoor, F.; van den Berg, H.H.; Vennema, H.; Duizer, E.; Koopmans, M.; de Roda Husman, A.M. Sources of hepatitis e virus genotype 3 in the netherlands. Emerg. Infect. Dis. 2009, 15, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Hakze-van der Honing, R.W.; van Coillie, E.; Antonis, A.F.G.; van der Poel, W.H.M. First isolation of hepatitis e virus genotype 4 in europe through swine surveillance in the netherlands and belgium. PLoS ONE 2011, 6, e22673. [Google Scholar] [CrossRef] [PubMed]
- De Deus, N.; Seminati, C.; Pina, S.; Mateu, E.; Martín, M.; Segalés, J. Detection of hepatitis e virus in liver, mesenteric lymph node, serum, bile and faeces of naturally infected pigs affected by different pathological conditions. Vet. Microbiol. 2007, 119, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Steyer, A.; Naglič, T.; Močilnik, T.; Poljšak-Prijatelj, M.; Poljak, M. Hepatitis e virus in domestic pigs and surface waters in slovenia: Prevalence and molecular characterization of a novel genotype 3 lineage. Infect. Genet. Evol. 2011, 11, 1732–1737. [Google Scholar] [CrossRef] [PubMed]
- Vasickova, P.; Psikal, I.; Widen, F.; Smitalova, R.; Bendova, J.; Pavlik, I.; Kralik, P. Detection and genetic characterisation of hepatitis e virus in czech pig production herds. Res. Vet. Sci. 2009, 87, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Widén, F.; Sundqvist, L.; Matyi-Toth, A.; Metreveli, G.; Belák, S.; Hallgren, G.; Norder, H. Molecular epidemiology of hepatitis e virus in humans, pigs and wild boars in sweden. Epidemiol. Infect. 2011, 139, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J. Recent advances in hepatitis e virus. J. Viral Hepat. 2010, 17, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.W.; Smith, M.M.; Guerra, M.E.; Huang, C.-C.C.; Bradley, D.W.; Fry, K.E.; Reyes, G.R. Hepatitis e virus (hev): Molecular cloning and sequencing of the full-length viral genome. Virology 1991, 185, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis e virus: Delineation of an additional group of positive-strand rna plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H. Hepatitis e virus cell culture models. Virus Res. 2011, 161, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Simmonds, P.; members of the International Committee on the Taxonomy of Viruses Hepeviridae Study Group; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; van der Poel, W.H.; Purdy, M.A. Consensus proposals for classification of the family hepeviridae. J. Gen. Virol. 2014, 95, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Purdy, M.A.; Khudyakov, Y.E. Evolutionary history and population dynamics of hepatitis e virus. PLoS ONE 2010, 5, e14376. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, C.; Hagedorn, C.H. Phylogenetic analysis of global hepatitis e virus sequences: Genetic diversity, subtypes and zoonosis. Rev. Med. Virol. 2006, 16, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Nguyen, D.; Fernandez, J.; Yun, K.Y.; Fry, K.E.; Bradley, D.W.; Tam, A.W.; Reyes, G.R. Molecular cloning and sequencing of the mexico isolate of hepatitis e virus (hev). Virology 1992, 191, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Van Cuyck-Gandré, H.; Zhang, H.Y.; Tsarev, S.A.; Clements, N.J.; Cohen, S.J.; Caudill, J.D.; Buisson, Y.; Coursaget, P.; Warren, R.L.; Longer, C.F. Characterization of hepatitis e virus (hev) from algeria and chad by partial genome sequence. J. Med. Virol. 1997, 53, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ling, R.; Erker, J.C.; Zhang, H.; Li, H.; Desai, S.; Mushahwar, I.K.; Harrison, T.J. A divergent genotype of hepatitis e virus in chinese patients with acute hepatitis. J. Gen. Virol. 1999, 80, 169–177. [Google Scholar] [PubMed]
- Kwo, P.Y.; Schlauder, G.G.; Carpenter, H.A.; Murphy, P.J.; Rosenblatt, J.E.; Dawson, G.J.; Mast, E.E.; Krawczynski, K.; Balan, V. Acute hepatitis e by a new isolate acquired in the united states. Mayo Clin. Proc. Mayo Clin. 1997, 72, 1133–1136. [Google Scholar] [CrossRef]
- Aggarwal, R.; Naik, S. Epidemiology of hepatitis e: Current status. J. Gastroenterol. Hepatol. 2009, 24, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Robert Koch-Institut. Available online: http://www.rki.de (accessed on 7 April 2015).
- Scharn, N.; Ganzenmueller, T.; Wenzel, J.J.; Dengler, R.; Heim, A.; Wegner, F. Guillain-barre syndrome associated with autochthonous infection by hepatitis e virus subgenotype 3c. Infection 2014, 42, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Manka, P.; Bechmann, L.P.; Coombes, J.D.; Thodou, V.; Schlattjan, M.; Kahraman, A.; Syn, W.K.; Saner, F.; Gerken, G.; Baba, H.; et al. Hepatitis e virus infection as a possible cause of acute liver failure in europe. Clin. Gastroenterol. Hepatol. 2015. [Google Scholar] [CrossRef]
- Takahashi, K.; Okamoto, H.; Abe, N.; Kawakami, M.; Matsuda, H.; Mochida, S.; Sakugawa, H.; Suginoshita, Y.; Watanabe, S.; Yamamoto, K.; et al. Virulent strain of hepatitis e virus genotype 3, japan. Emerg. Infect. Dis. 2009, 15, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, S.; Frickmann, H.; Gabriel, M.; Schmitz, N.; Gunther, S.; Schmidt-Chanasit, J. Fatal course of an autochthonous hepatitis e virus infection in a patient with leukemia in germany. Infection 2012, 40, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Zehender, G.; Ebranati, E.; Lai, A.; Luzzago, C.; Paladini, S.; Tagliacarne, C.; Galli, C.; Galli, M.; Ciccozzi, M.; Zanetti, A.R.; et al. Phylogeography and phylodynamics of european genotype 3 hepatitis e virus. Infect. Genet. Evol. 2014, 25, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Okano, H.; Kobayashi, M.; Ito, K.; Ohmori, S.; Nomura, T.; Kato, H.; Ayada, M.; Nakano, Y.; Akachi, S.; et al. Molecular epidemiology and genetic history of european-type genotype 3 hepatitis e virus indigenized in the central region of japan. Infect. Genet. Evol. 2012, 12, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Takahashi, K.; Pybus, O.G.; Hashimoto, N.; Kato, H.; Okano, H.; Kobayashi, M.; Fujita, N.; Shiraki, K.; Takei, Y.; et al. New findings regarding the epidemic history and population dynamics of japan-indigenous genotype 3 hepatitis e virus inferred by molecular evolution. Liver Int. 2012, 32, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Takahashi, K.; Arai, M.; Okano, H.; Kato, H.; Ayada, M.; Okamoto, H.; Mishiro, S. Identification of european-type hepatitis e virus subtype 3e isolates in Japanese wild boars: Molecular tracing of hev from swine to wild boars. Infect. Genet. Evol. 2013, 18, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Borentain, P.; Motte, A.; Lagrange, X.; Kaba, M.; Henry, M.; Tamalet, C.; Gerolami, R. First human cases of hepatitis e infection with genotype 3c strains. J. Clin. Virol. 2007, 40, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Dalton, H.R.; Abravanel, F.; Izopet, J. Hepatitis e virus infection. Clin. Microbiol. Rev. 2014, 27, 116–138. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Depner, K.; Schirrmeier, H.; Beer, M. A universal heterologous internal control system for duplex real-time rt-pcr assays used in a detection system for pestiviruses. J. Virol. Methods 2006, 136, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. Bioedit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/nt. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Schlosser, J.; Eiden, M.; Vina-Rodriguez, A.; Fast, C.; Dremsek, P.; Lange, E.; Ulrich, R.G.; Groschup, M.H. Natural and experimental hepatitis e virus genotype 3-infection in european wild boar is transmissible to domestic pigs. Vet. Res. 2014, 45, e121. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Norder, H.; Sundqvist, L.; Magnusson, L.; Ostergaard Breum, S.; Löfdahl, M.; Larsen, L.E.; Hjulsager, C.K.; Magnius, L.; Böttiger, B.E.; Widen, F. Endemic hepatitis e in two nordic countries. Eurosurveillance 2009, 14, 1–9. [Google Scholar]
- Jothikumar, N.; Cromeans, T.L.; Robertson, B.H.; Meng, X.J.; Hill, V.R. A broadly reactive one-step real-time RT-PCR assay for rapid and sensitive detection of hepatitis e virus. J. Virol. Methods 2006, 131, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Williams, B.L.; King, N.; Carroll, S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Yoo, D. Genetic characterization and sequence heterogeneity of a canadian isolate of swine hepatitis e virus. J. Clin. Microbiol. 2002, 40, 4021–4029. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Bukh, J.; Combet, C.; Deleage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspe, G.; Kuiken, C.; Maertens, G.; et al. Consensus proposals for a unified system of nomenclature of hepatitis c virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Plenge-Bönig, A.; Hess, M.; Ulrich, R.G.; Reetz, J.; Schielke, A. Detection of a novel hepatitis e-like virus in faeces of wild rats using a nested broad-spectrum RT-PCR. J. Gen. Virol. 2010, 91, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Purdy, M.A. Evolution of the hepatitis e virus polyproline region: Order from disorder. J. Virol. 2012, 86, 10186–10193. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Mansuy, J.M.; Rostaing, L.; Kamar, N.; Izopet, J. Characterization of the polyproline region of the hepatitis e virus in immunocompromised patients. J. Virol. 2014, 88, 12017–12025. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Computational Molecular Evolution; Oxford University Press: Oxford, UK, 2006; pp. xvi, p. 357. [Google Scholar]
- Wichmann, O.; Schimanski, S.; Koch, J.; Kohler, M.; Rothe, C.; Plentz, A.; Jilg, W.; Stark, K. Phylogenetic and case-control study on hepatitis e virus infection in germany. J. Infect. Dis. 2008, 198, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Lopes Dos Santos, D.R.; Lewis-Ximenez, L.L.; da Silva, M.F.; de Sousa, P.S.; Gaspar, A.M.; Pinto, M.A. First report of a human autochthonous hepatitis e virus infection in brazil. J. Clin. Virol. 2010, 47, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Legrand-Abravanel, F.; Mansuy, J.M.; Dubois, M.; Kamar, N.; Peron, J.M.; Rostaing, L.; Izopet, J. Hepatitis e virus genotype 3 diversity, france. Emerg. Infect. Dis. 2009, 15, 110–114. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vina-Rodriguez, A.; Schlosser, J.; Becher, D.; Kaden, V.; Groschup, M.H.; Eiden, M. Hepatitis E Virus Genotype 3 Diversity: Phylogenetic Analysis and Presence of Subtype 3b in Wild Boar in Europe. Viruses 2015, 7, 2704-2726. https://doi.org/10.3390/v7052704
Vina-Rodriguez A, Schlosser J, Becher D, Kaden V, Groschup MH, Eiden M. Hepatitis E Virus Genotype 3 Diversity: Phylogenetic Analysis and Presence of Subtype 3b in Wild Boar in Europe. Viruses. 2015; 7(5):2704-2726. https://doi.org/10.3390/v7052704
Chicago/Turabian StyleVina-Rodriguez, Ariel, Josephine Schlosser, Dietmar Becher, Volker Kaden, Martin H. Groschup, and Martin Eiden. 2015. "Hepatitis E Virus Genotype 3 Diversity: Phylogenetic Analysis and Presence of Subtype 3b in Wild Boar in Europe" Viruses 7, no. 5: 2704-2726. https://doi.org/10.3390/v7052704
APA StyleVina-Rodriguez, A., Schlosser, J., Becher, D., Kaden, V., Groschup, M. H., & Eiden, M. (2015). Hepatitis E Virus Genotype 3 Diversity: Phylogenetic Analysis and Presence of Subtype 3b in Wild Boar in Europe. Viruses, 7(5), 2704-2726. https://doi.org/10.3390/v7052704