
The Emerging Roles of Viroporins in ER Stress Response and Autophagy Induction during Virus Infection


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
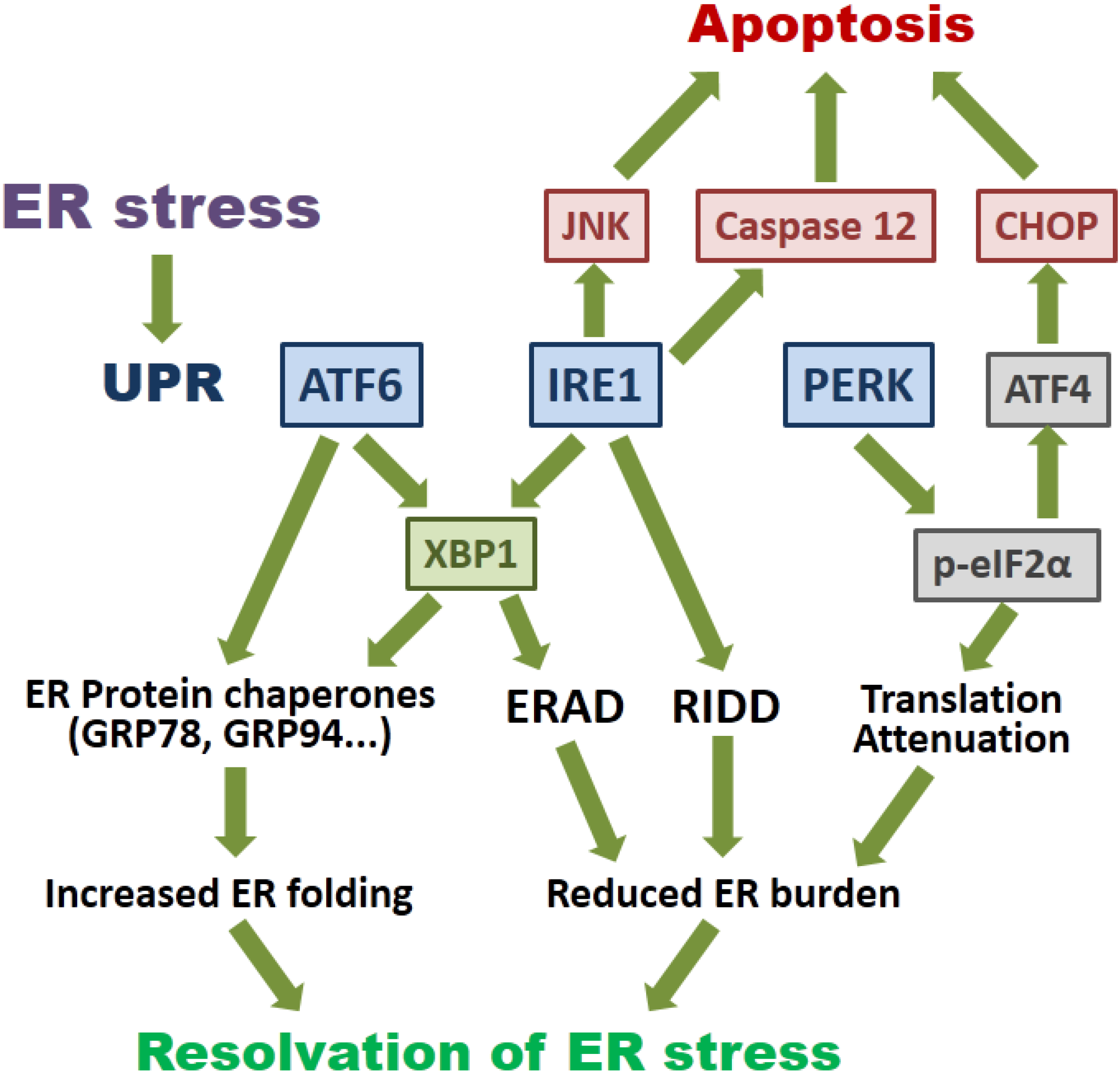
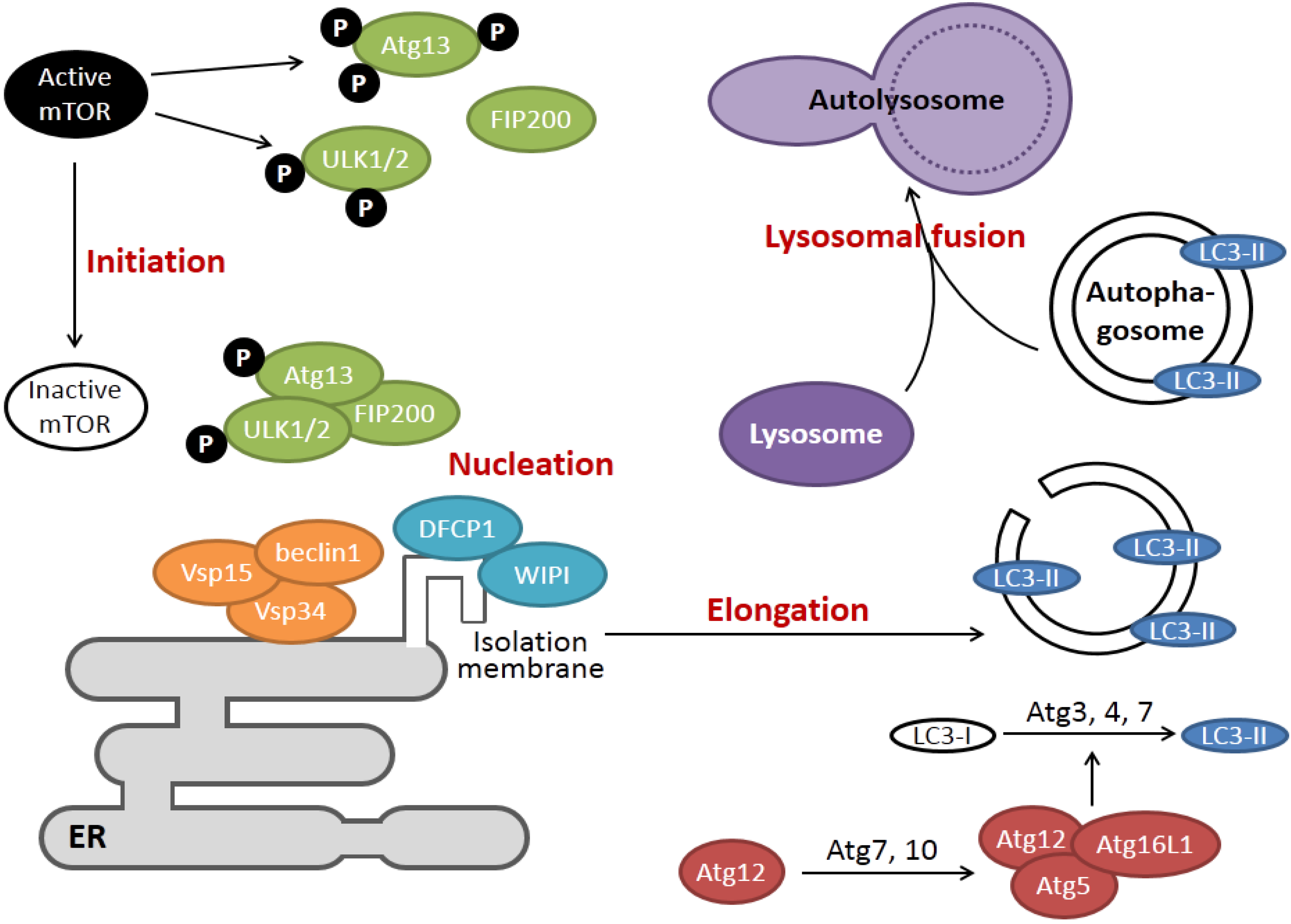
2. Overview of the Signaling Pathways of ER Stress and Autophagy


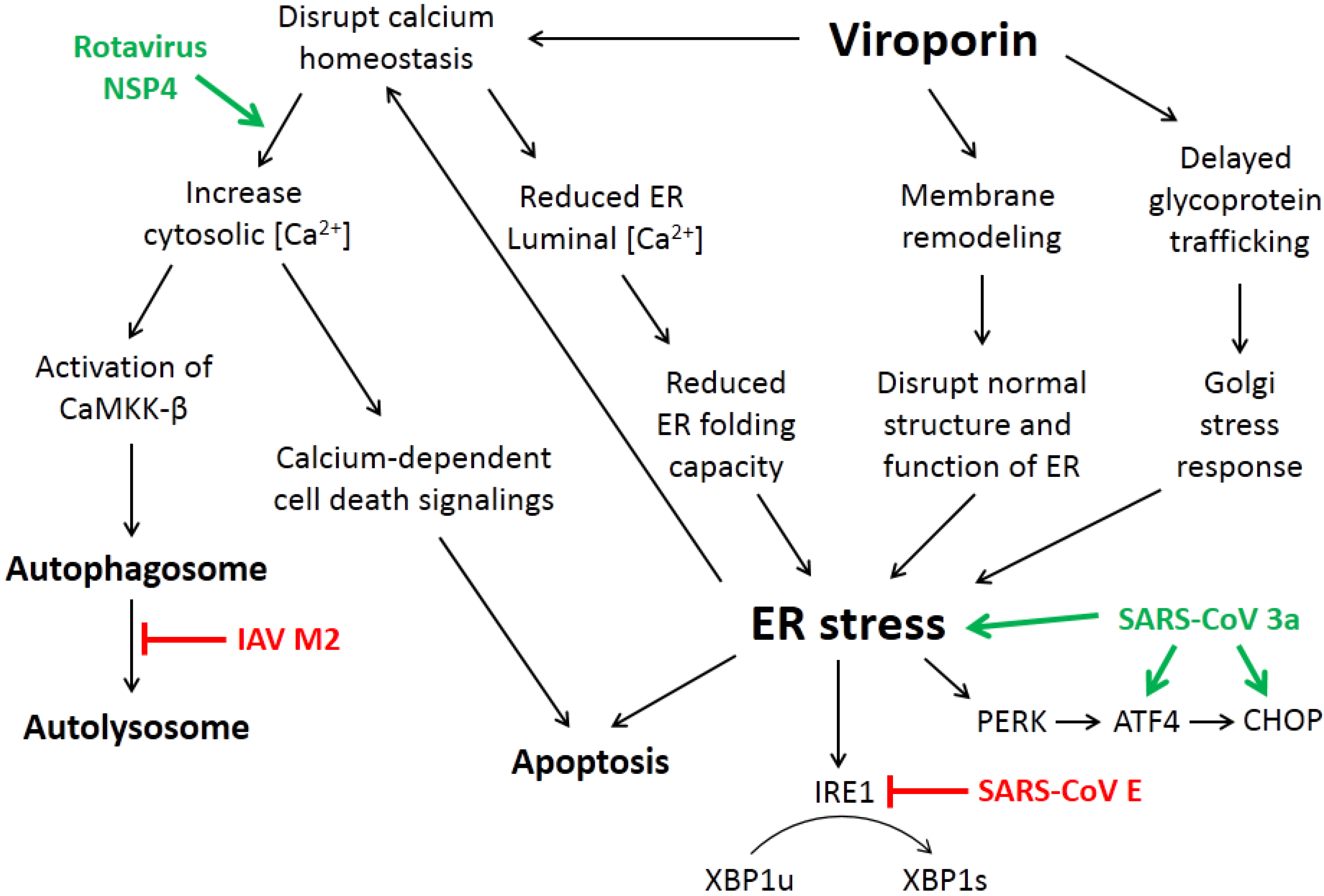
3. How Viroporin Might Modulate ER Stress and Autophagy

3.1. ER Calcium Homeostasis
3.2. Membrane Rearrangement
3.3. Intracellular Trafficking of Glycoproteins
4. Current Studies of Viroporins Modulating ER Stress and Autophagy
4.2. The SARS-CoV Accessory Protein 3a Induces ER Stress and Apoptosis
4.3. Rotavirus NSP4 Hijacks Autophagy for Replication
4.4. Influenza A Virus M2 Protein and the Subversion of Autophagy
4.5. Other Small Membrane-Associated Viral Proteins Modulating ER Stress Response and Autophagy Induction without Confirmed Ion Channel Activities
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Aldabe, R.; Barco, A.; Carrasco, L. Membrane permeabilization by poliovirus proteins 2B and 2BC. J. Biol. Chem. 1996, 271, 23134–23137. [Google Scholar] [PubMed]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.J.; Wolstenholme, A.J.; Skehel, J.J.; Smith, M.H. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985, 4, 3021–3024. [Google Scholar] [PubMed]
- Helenius, A. Unpacking the incoming influenza virus. Cell 1992, 69, 577–578. [Google Scholar] [CrossRef]
- Sugrue, R.J.; Bahadur, G.; Zambon, M.C.; Hall-Smith, M.; Douglas, A.R.; Hay, A.J. Specific structural alteration of the influenza haemagglutinin by amantadine. EMBO J. 1990, 9, 3469–3476. [Google Scholar] [PubMed]
- Steinmann, E.; Penin, F.; Kallis, S.; Patel, A.H.; Bartenschlager, R.; Pietschmann, T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007, 3, e103. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Castelló, A.; Carrasco, L. Viroporins from RNA viruses induce caspase-dependent apoptosis. Cell. Microbiol. 2008, 10, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Magadán, J.G.; Bonifacino, J.S. Transmembrane domain determinants of CD4 downregulation by HIV-1 Vpu. J. Virol. 2012, 86, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, Y.; Fujita, H.; Kinomoto, M.; Kaneko, K.; Ishizaka, Y.; Tanaka, Y.; Sata, T.; Tokunaga, K. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 2009, 284, 35060–35072. [Google Scholar] [CrossRef] [PubMed]
- Magadán, J.G.; Pérez-Victoria, F.J.; Sougrat, R.; Ye, Y.; Strebel, K.; Bonifacino, J.S. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 2010, 6, e1000869. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Klappa, P.; Kietzmann, T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef] [PubMed]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; de Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Alvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.-J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS ONE 2009, 4, e8342. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Hyser, J.M.; Utama, B.; Estes, M.K. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-β signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. USA 2012, 109, E3405–E3413. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yoshida, H.; Kokame, K.; Kaufman, R.J.; Mori, K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 2004, 136, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Deretic, V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007, 7, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Jheng, J.-R.; Ho, J.-Y.; Horng, J.-T. ER stress, autophagy, and RNA viruses. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Huang, M.; Liu, D.X. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus-host interactions. Virus Res. 2014, 194, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Watowich, S.S.; Morimoto, R.I.; Lamb, R.A. Flux of the paramyxovirus hemagglutinin-neuraminidase glycoprotein through the endoplasmic reticulum activates transcription of the GRP78-BiP gene. J. Virol. 1991, 65, 3590–3597. [Google Scholar] [PubMed]
- Chan, C.-P.; Siu, K.-L.; Chin, K.-T.; Yuen, K.-Y.; Zheng, B.; Jin, D.-Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Collinson-Pautz, M.R.; Utama, B.; Estes, M.K. Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. mBio 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-C.; Sun, S.-Q.; Sun, D.-H.; Wei, Y.-Q.; Xu, J.; Huang, M.; Liu, X.-T.; Liu, Z.-X.; Luo, J.-X.; Yin, H.; et al. Viroporin activity and membrane topology of classic swine fever virus p7 protein. Int. J. Biochem. Cell Biol. 2013, 45, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Hammadi, M.; Oulidi, A.; Gackière, F.; Katsogiannou, M.; Slomianny, C.; Roudbaraki, M.; Dewailly, E.; Delcourt, P.; Lepage, G.; Lotteau, S.; et al. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: Involvement of GRP78. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Ao, D.; Guo, H.-C.; Sun, S.-Q.; Sun, D.-H.; Fung, T.S.; Wei, Y.-Q.; Han, S.-C.; Yao, X.-P.; Cao, S.-Z.; Liu, D.X.; et al. Viroporin Activity of the Foot-and-Mouth disease virus non-structural 2B protein. PLoS ONE 2015, 10, e0125828. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- David-Ferreira, J.F.; Manaker, R.A. An electron microscope study of the development of a mouse hepatitis virus in tissue culture cells. J. Cell Biol. 1965, 24, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Dales, S.; Eggers, H.J.; Tamm, I.; Palade, G.E. Electron microscopic study of the formation of poliovirus. Virology 1965, 26, 379–389. [Google Scholar] [CrossRef]
- Knoops, K.; Kikkert, M.; van den Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replicationi supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Suhy, D.A.; Giddings, T.H.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef] [PubMed]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Calì, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.M.; Molinari, M. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived erad regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Doedens, J.R.; Kirkegaard, K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995, 14, 894–907. [Google Scholar] [PubMed]
- De Jong, A.S.; Visch, H.-J.; de Mattia, F.; van Dommelen, M.M.; Swarts, H.G.; Luyten, T.; Callewaert, G.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. The coxsackievirus 2B protein increases efflux of ions from the endoplasmic reticulum and Golgi, thereby inhibiting protein trafficking through the Golgi. J. Biol. Chem. 2006, 281, 14144–14150. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, T.; Leser, G.P.; Lamb, R.A. The ion channel activity of the influenza virus M2 protein affects transport through the Golgi apparatus. J. Cell Biol. 1996, 133, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.L.; Griffin, S.; Rowlands, D.; Harris, M.; Yi, M.; Lemon, S.M.; Weinman, S.A. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 2010, 6, e1001087. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Yoshida, H. Organelle autoregulation-stress responses in the ER, Golgi, mitochondria and lysosome. J. Biochem. 2015, 157, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Inglis, S.C. Association of the infectious bronchitis virus 3c protein with the virion envelope. Virology 1991, 185, 911–917. [Google Scholar] [CrossRef]
- Godet, M.; L’Haridon, R.; Vautherot, J.F.; Laude, H. TGEV corona virus ORF4 encodes a membrane protein that is incorporated into virions. Virology 1992, 188, 666–675. [Google Scholar] [CrossRef]
- Yuan, Q.; Liao, Y.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical evidence for the presence of mixed membrane topologies of the severe acute respiratory syndrome coronavirus envelope protein expressed in mammalian cells. FEBS Lett. 2006, 580, 3192–3200. [Google Scholar] [CrossRef] [PubMed]
- Corse, E.; Machamer, C.E. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J. Virol. 2000, 74, 4319–4326. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Yuan, Q.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology 2006, 349, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.R.; Lin, L.D.; Machamer, C.E. Identification of a Golgi complex-targeting signal in the cytoplasmic tail of the severe acute respiratory syndrome coronavirus envelope protein. J. Virol. 2011, 85, 5794–5803. [Google Scholar] [CrossRef] [PubMed]
- Raamsman, M.J.; Locker, J.K.; de Hooge, A.; de Vries, A.A.; Griffiths, G.; Vennema, H.; Rottier, P.J. Characterization of the coronavirus mouse hepatitis virus strain A59 small membrane protein E. J. Virol. 2000, 74, 2333–2342. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Dediego, M.L.; Alvarez, E.; Jiménez-Guardeño, J.M.; Regla-Nava, J.A.; Llorente, M.; Kremer, L.; Shuo, S.; Enjuanes, L. Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology 2011, 415, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Maeda, J.; Repass, J.F.; Maeda, A.; Makino, S. Membrane topology of coronavirus E protein. Virology 2001, 281, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Surya, W.; Claudine, S.; Torres, J. Structure of a conserved Golgi complex-targeting signal in coronavirus envelope proteins. J. Biol. Chem. 2014, 289, 12535–12549. [Google Scholar] [CrossRef] [PubMed]
- Surya, W.; Li, Y.; Verdià-Bàguena, C.; Aguilella, V.M.; Torres, J. MERS coronavirus envelope protein has a single transmembrane domain that forms pentameric ion channels. Virus Res. 2015, 201, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Corse, E.; Machamer, C.E. The cytoplasmic tail of infectious bronchitis virus E protein directs Golgi targeting. J. Virol. 2002, 76, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Boscarino, J.A.; Logan, H.L.; Lacny, J.J.; Gallagher, T.M. Envelope protein palmitoylations are crucial for murine coronavirus assembly. J. Virol. 2008, 82, 2989–2999. [Google Scholar] [CrossRef] [PubMed]
- Arbely, E.; Khattari, Z.; Brotons, G.; Akkawi, M.; Salditt, T.; Arkin, I.T. A highly unusual palindromic transmembrane helical hairpin formed by SARS coronavirus E protein. J. Mol. Biol. 2004, 341, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.P.; Liu, D.X. The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre-Golgi compartments and physical interaction between the envelope and membrane proteins. J. Biol. Chem. 2001, 276, 17515–17523. [Google Scholar] [CrossRef] [PubMed]
- Vennema, H.; Godeke, G.J.; Rossen, J.W.; Voorhout, W.F.; Horzinek, M.C.; Opstelten, D.J.; Rottier, P.J. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996, 15, 2020–2028. [Google Scholar] [PubMed]
- Bos, E.C.; Luytjes, W.; van der Meulen, H.V.; Koerten, H.K.; Spaan, W.J. The production of recombinant infectious DI-particles of a murine coronavirus in the absence of helper virus. Virology 1996, 218, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Masters, P.S. The small envelope protein E is not essential for murine coronavirus replication. J. Virol. 2003, 77, 4597–4608. [Google Scholar] [CrossRef] [PubMed]
- Ortego, J.; Ceriani, J.E.; Patiño, C.; Plana, J.; Enjuanes, L. Absence of E protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 2007, 368, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Almazán, F.; DeDiego, M.L.; Sola, I.; Zuñiga, S.; Nieto-Torres, J.L.; Marquez-Jurado, S.; Andrés, G.; Enjuanes, L. Engineering a replication-competent, propagation-defective Middle East respiratory syndrome coronavirus as a vaccine candidate. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Yuan, Q.; Liao, Y. Coronavirus envelope protein: A small membrane protein with multiple functions. Cell. Mol. Life Sci. 2007, 64, 2043–2048. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; McKinlay, C.; Gage, P.; Ewart, G. SARS coronavirus E protein forms cation-selective ion channels. Virology 2004, 330, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; Gage, P.; Ewart, G. Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology 2006, 353, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Verdiá-Báguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; DeDiego, M.L.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology 2012, 432, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Lescar, J.; Tam, J.P.; Liu, D.X. Expression of SARS-coronavirus envelope protein in Escherichia coli cells alters membrane permeability. Biochem. Biophys. Res. Commun. 2004, 325, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; García Mde, J.; Sanz, M.A.; Carrasco, L. Viroporin activity of murine hepatitis virus E protein. FEBS Lett. 2005, 579, 3607–3612. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.; Tan, E.; Parthasarathy, K.; Lin, X.; Jiang, F.L.; Yu, D.; Vararattanavech, A.; Soong, T.W.; Liu, D.X.; Torres, J. Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog. 2009, 5, e1000511. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Parthasarathy, K.; Lin, X.; Saravanan, R.; Kukol, A.; Liu, D.X. Model of a putative pore: The pentameric α-helical bundle of SARS coronavirus E protein in lipid bilayers. Biophys. J. 2006, 91, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Chen, C.-J.; Yu, X.; Leibowitz, J.L.; Makino, S. Induction of apoptosis in murine coronavirus-infected cultured cells and demonstration of E protein as an apoptosis inducer. J. Virol. 1999, 73, 7853–7859. [Google Scholar] [PubMed]
- Yang, Y.; Xiong, Z.; Zhang, S.; Yan, Y.; Nguyen, J.; Ng, B.; Lu, H.; Brendese, J.; Yang, F.; Wang, H.; et al. Bcl-xL inhibits T-cell apoptosis induced by expression of SARS coronavirus E protein in the absence of growth factors. Biochem. J. 2005, 392, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Ruch, T.R.; Machamer, C.E. A Single Polar Residue and Distinct Membrane Topologies impact the function of the infectious bronchitis coronavirus E protein. PLoS Pathog. 2012, 8, e1002674. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jiménez-Guardeño, J.M.; Regla-Nava, J.A.; Álvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liao, Y.; Liu, D.X. The Endoplasmic reticulum stress sensor IRE1α protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-J.; Chen, Y.-C.; Hsiao, C.-H.; Kuo, T.-C.; Chang, S.C.; Lu, C.-Y.; Wei, W.-C.; Lee, C.-H.; Huang, L.-M.; Chang, M.-F.; et al. Identification of a novel protein 3a from severe acute respiratory syndrome coronavirus. FEBS Lett. 2004, 565, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Yount, B.; Roberts, R.S.; Sims, A.C.; Deming, D.; Frieman, M.B.; Sparks, J.; Denison, M.R.; Davis, N.; Baric, R.S. Severe Acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J. Virol. 2005, 79, 14909–14922. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Fung, T.S.; Chong, K.K.-L.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014, 109, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wong, S.-M.; Liu, D.X. Identification of Hepta- and Octo-Uridine stretches as sole signals for programmed +1 and −1 ribosomal frameshifting during translation of SARS-CoV ORF 3a variants. Nucleic Acids Res. 2006, 34, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Mossel, E.C.; Narayanan, K.; Popov, V.L.; Huang, C.; Inoue, T.; Peters, C.J.; Makino, S. Severe acute respiratory syndrome coronavirus 3a protein is a viral structural protein. J. Virol. 2005, 79, 3182–3186. [Google Scholar] [CrossRef] [PubMed]
- Oostra, M.; de Haan, C.A.; de Groot, R.J.; Rottier, P.J. Glycosylation of the severe acute respiratory syndrome coronavirus triple-spanning membrane proteins 3a and M. J. Virol. 2006, 80, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-J.; Teng, E.; Shen, S.; Tan, T.H.P.; Goh, P.-Y.; Fielding, B.C.; Ooi, E.-E.; Tan, H.-C.; Lim, S.G.; Hong, W. A novel severe acute respiratory syndrome coronavirus protein, U274, is transported to the cell surface and undergoes endocytosis. J. Virol. 2004, 78, 6723–6734. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zheng, B.-J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.L.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [PubMed]
- Law, P.T.W.; Wong, C.-H.; Au, T.C.C.; Chuck, C.-P.; Kong, S.-K.; Chan, P.K.S.; To, K.-F.; Lo, A.W.I.; Chan, J.Y.W.; Suen, Y.-K.; et al. The 3a protein of severe acute respiratory syndrome-associated coronavirus induces apoptosis in Vero E6 cells. J. Gen. Virol. 2005, 86, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.A.; Chen, Y.; Chan, C.M.; Chan, C.S.M.; Chan, P.K.S.; Chui, Y.L.; Fung, K.P.; Waye, M.M.Y.; Tsui, S.K.W.; Chan, H.Y.E. In vivo functional characterization of the SARS-Coronavirus 3a protein in Drosophila. Biochem. Biophys. Res. Commun. 2005, 337, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Freundt, E.C.; Yu, L.; Goldsmith, C.S.; Welsh, S.; Cheng, A.; Yount, B.; Liu, W.; Frieman, M.B.; Buchholz, U.J.; Screaton, G.R.; et al. The open reading frame 3a protein of severe acute respiratory syndrome-associated coronavirus promotes membrane rearrangement and cell death. J. Virol. 2010, 84, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Ball, J.M.; Tian, P.; Zeng, C.Q.; Morris, A.P.; Estes, M.K. Age-dependent diarrhea induced by a rotaviral nonstructural glycoprotein. Science 1996, 272, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.D.; Nodelman, I.M.; Levy, O.; Lin, S.L.; Tian, P.; Zamb, T.J.; Udem, S.A.; Venkataraghavan, B.; Schutt, C.E. Crystal structure of the oligomerization domain of NSP4 from rotavirus reveals a core metal-binding site. J. Mol. Biol. 2000, 304, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Sastri, N.P.; Viskovska, M.; Hyser, J.M.; Tanner, M.R.; Horton, L.B.; Sankaran, B.; Prasad, B.V.V.; Estes, M.K. Structural plasticity of the coiled-coil domain of rotavirus NSP4. J. Virol. 2014, 88, 13602–13612. [Google Scholar] [CrossRef] [PubMed]
- Michelangeli, F.; Ruiz, M.C.; del Castillo, J.R.; Ludert, J.E.; Liprandi, F. Effect of rotavirus infection on intracellular calcium homeostasis in cultured cells. Virology 1991, 181, 520–527. [Google Scholar] [CrossRef]
- Hyser, J.M.; Utama, B.; Crawford, S.E.; Broughman, J.R.; Estes, M.K. Activation of the endoplasmic reticulum calcium sensor stim1 and store-operated calcium entry by rotavirus requires NSP4 viroporin activity. J. Virol. 2013, 87, 13579–13588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zeng, C.Q.; Morris, A.P.; Estes, M.K. A functional NSP4 enterotoxin peptide secreted from rotavirus-infected cells. J. Virol. 2000, 74, 11663–11670. [Google Scholar] [CrossRef] [PubMed]
- Didsbury, A.; Wang, C.; Verdon, D.; Sewell, M.A.; McIntosh, J.D.; Taylor, J.A. Rotavirus NSP4 is secreted from infected cells as an oligomeric lipoprotein and binds to glycosaminoglycans on the surface of non-infected cells. Virol. J. 2011, 8, e551. [Google Scholar] [CrossRef] [PubMed]
- Berkova, Z.; Crawford, S.E.; Trugnan, G.; Yoshimori, T.; Morris, A.P.; Estes, M.K. Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J. Virol. 2006, 80, 6061–6071. [Google Scholar] [CrossRef] [PubMed]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Beale, R.; Wise, H.; Stuart, A.; Ravenhill, B.J.; Digard, P.; Randow, F. A LC3-interacting motif in the influenza a virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 2014, 15, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.E.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [PubMed]
- Roosendaal, J.; Westaway, E.G.; Khromykh, A.; Mackenzie, J.M. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol. 2006, 80, 4623–4632. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Bühler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef] [PubMed]
- Kaufusi, P.H.; Kelley, J.F.; Yanagihara, R.; Nerurkar, V.R. Induction of endoplasmic reticulum-derived replication-competent membrane structures by West Nile virus non-structural protein 4B. PLoS ONE 2014, 9, e84040. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, R.L.; Mackenzie, J.M. West Nile Virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 2011, 85, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, R.L.; Mackenzie, J.M. ATF6 signaling is required for efficient West Nile Virus replication by promoting cell survival and inhibition of innate immune responses. J. Virol. 2013, 87, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ye, L.; Yu, X.; Xu, B.; Li, K.; Zhu, X.; Liu, H.; Wu, X.; Kong, L. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-κB activation. Virology 2009, 391, 257–264. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, A.-B.; Martín-Acebes, M.A.; Saiz, J.-C. Amino acid substitutions in the non-structural proteins 4A or 4B modulate the induction of autophagy in West Nile virus infected cells independently of the activation of the unfolded protein response. Front. Microbiol. 2015, 5, e797. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, A.-B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.-C.; Martín-Acebes, M.A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, e266. [Google Scholar] [CrossRef] [PubMed]
- Oostra, M.; Hagemeijer, M.C.; van Gent, M.; Bekker, C.P.J.; te Lintelo, E.G.; Rottier, P.J.M.; de Haan, C.A.M. Topology and membrane anchoring of the coronavirus replication complex: Not all hydrophobic domains of NSP3 and NSP6 are Membrane Spanning. J. Virol. 2008, 82, 12392–12405. [Google Scholar] [CrossRef] [PubMed]
- Baliji, S.; Cammer, S.A.; Sobral, B.; Baker, S.C. Detection of nonstructural protein 6 in murine coronavirus-infected cells and analysis of the transmembrane topology by using bioinformatics and molecular approaches. J. Virol. 2009, 83, 6957–6962. [Google Scholar] [CrossRef] [PubMed]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Lundin, A.; Dijkman, R.; Bergström, T.; Kann, N.; Adamiak, B.; Hannoun, C.; Kindler, E.; Jónsdóttir, H.R.; Muth, D.; Kint, J.; et al. Targeting membrane-bound viral rna synthesis reveals potent inhibition of diverse coronaviruses including the middle east respiratory syndrome virus. PLoS Pathog. 2014, 10, e1004166. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus NSP6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.N.; Denison, M.R.; Virgin, H.W. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007, 3, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A. A new paradigm: Innate immune sensing of viruses via the unfolded protein response. Front. Microbiol. 2014, 5, e222. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fung, T.S.; Torres, J.; Liu, D.X. The Emerging Roles of Viroporins in ER Stress Response and Autophagy Induction during Virus Infection. Viruses 2015, 7, 2834-2857. https://doi.org/10.3390/v7062749
Fung TS, Torres J, Liu DX. The Emerging Roles of Viroporins in ER Stress Response and Autophagy Induction during Virus Infection. Viruses. 2015; 7(6):2834-2857. https://doi.org/10.3390/v7062749
Chicago/Turabian StyleFung, To Sing, Jaume Torres, and Ding Xiang Liu. 2015. "The Emerging Roles of Viroporins in ER Stress Response and Autophagy Induction during Virus Infection" Viruses 7, no. 6: 2834-2857. https://doi.org/10.3390/v7062749
APA StyleFung, T. S., Torres, J., & Liu, D. X. (2015). The Emerging Roles of Viroporins in ER Stress Response and Autophagy Induction during Virus Infection. Viruses, 7(6), 2834-2857. https://doi.org/10.3390/v7062749