
Hydrogen Peroxide Induce Human Cytomegalovirus Replication through the Activation of p38-MAPK Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Chemical Reagents and Antibodies
2.2. Plasmids
2.3. Virus Preparation, Titration and Infection
2.4. DCF Staining
2.5. Detection of Cellular Catalase Activity and Intracellular H2O2 Level
2.6. Luciferase Assays
2.7. Real-Time PCR
2.8. qRT-PCR
2.9. Western Blot Analysis
2.10. Animal Studies
2.11. Statistical Analysis
3. Results
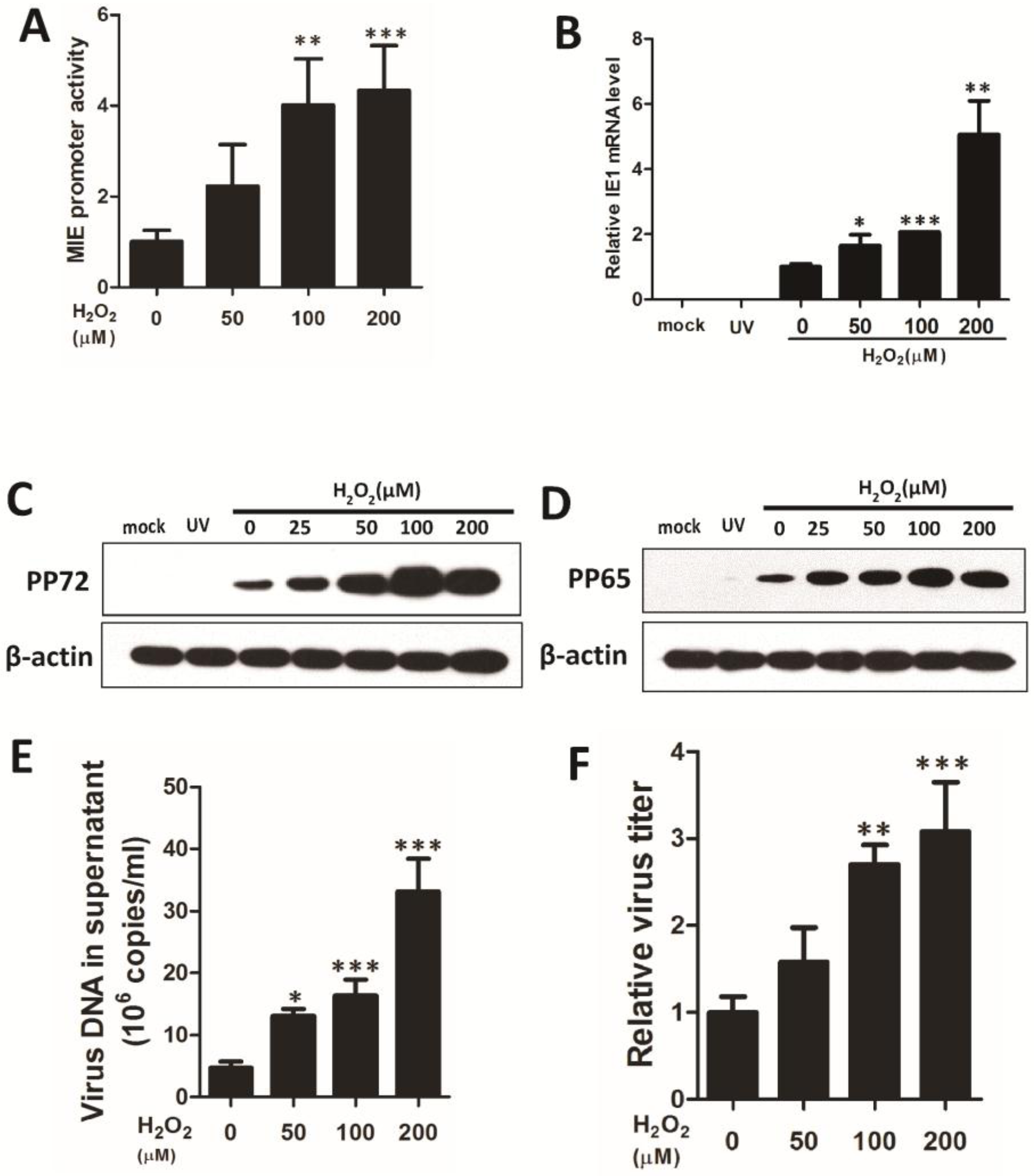
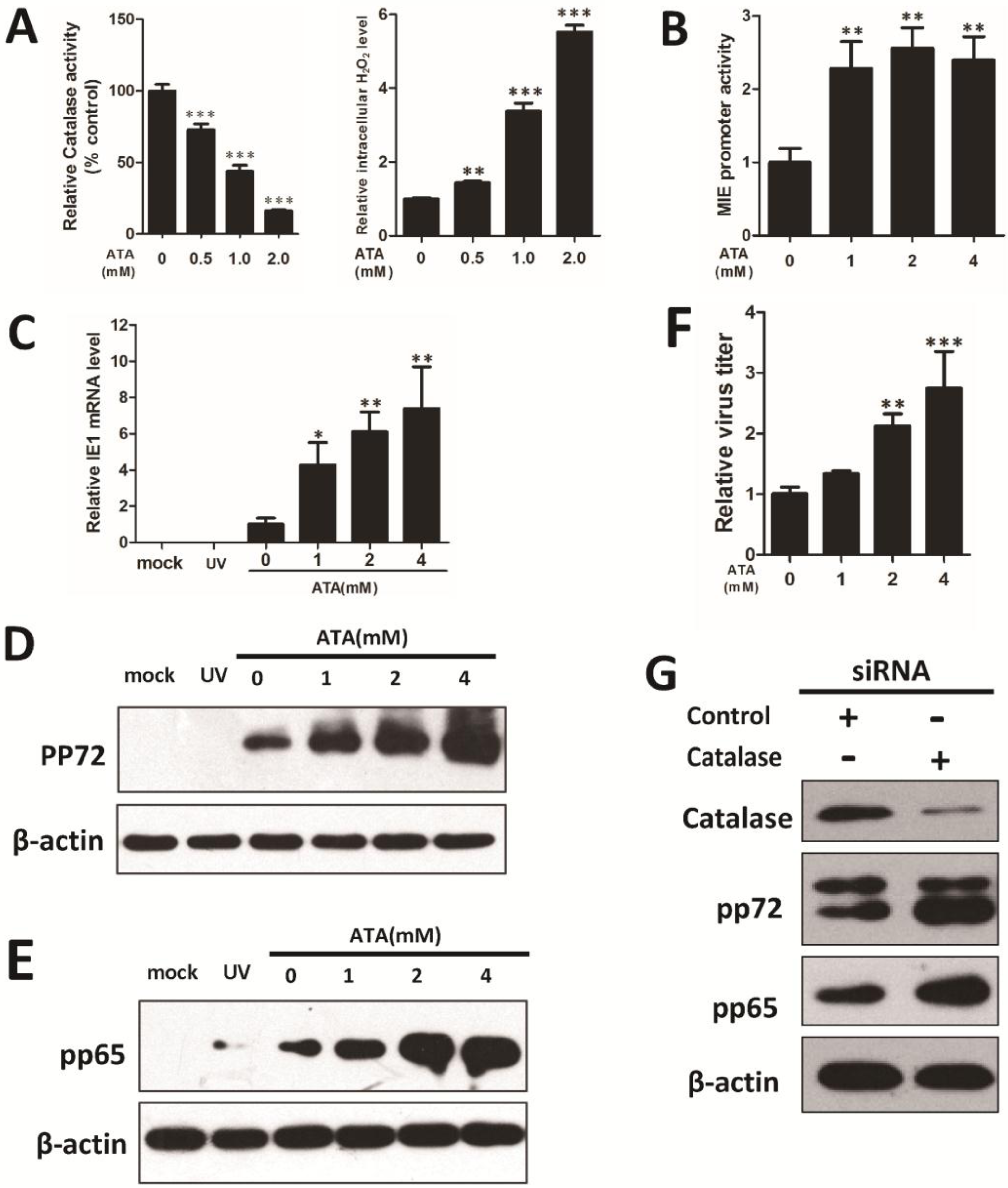
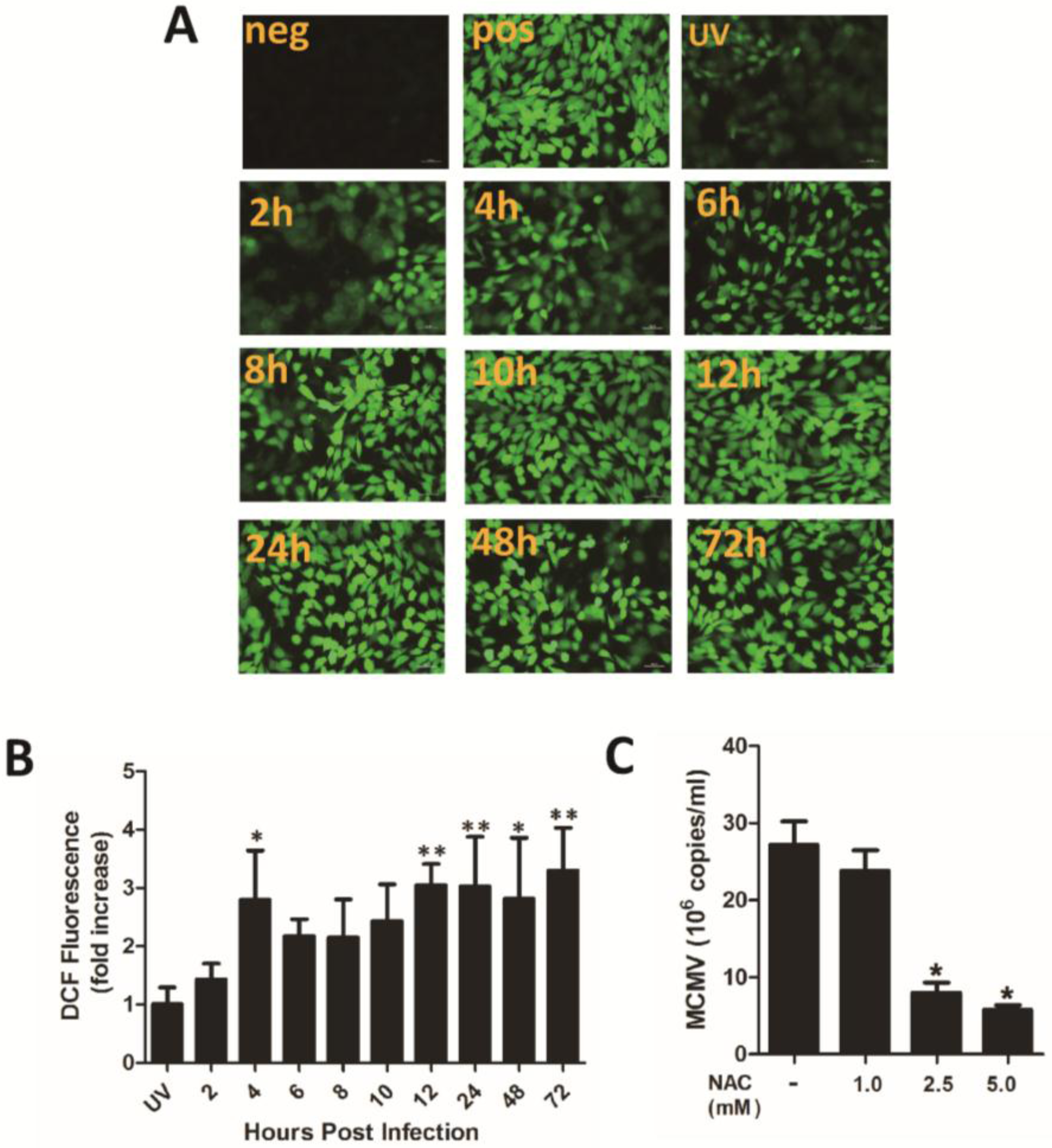
3.1. ROS Enhance HCMV Replication through Paracrine and Autocrine Mechanisms



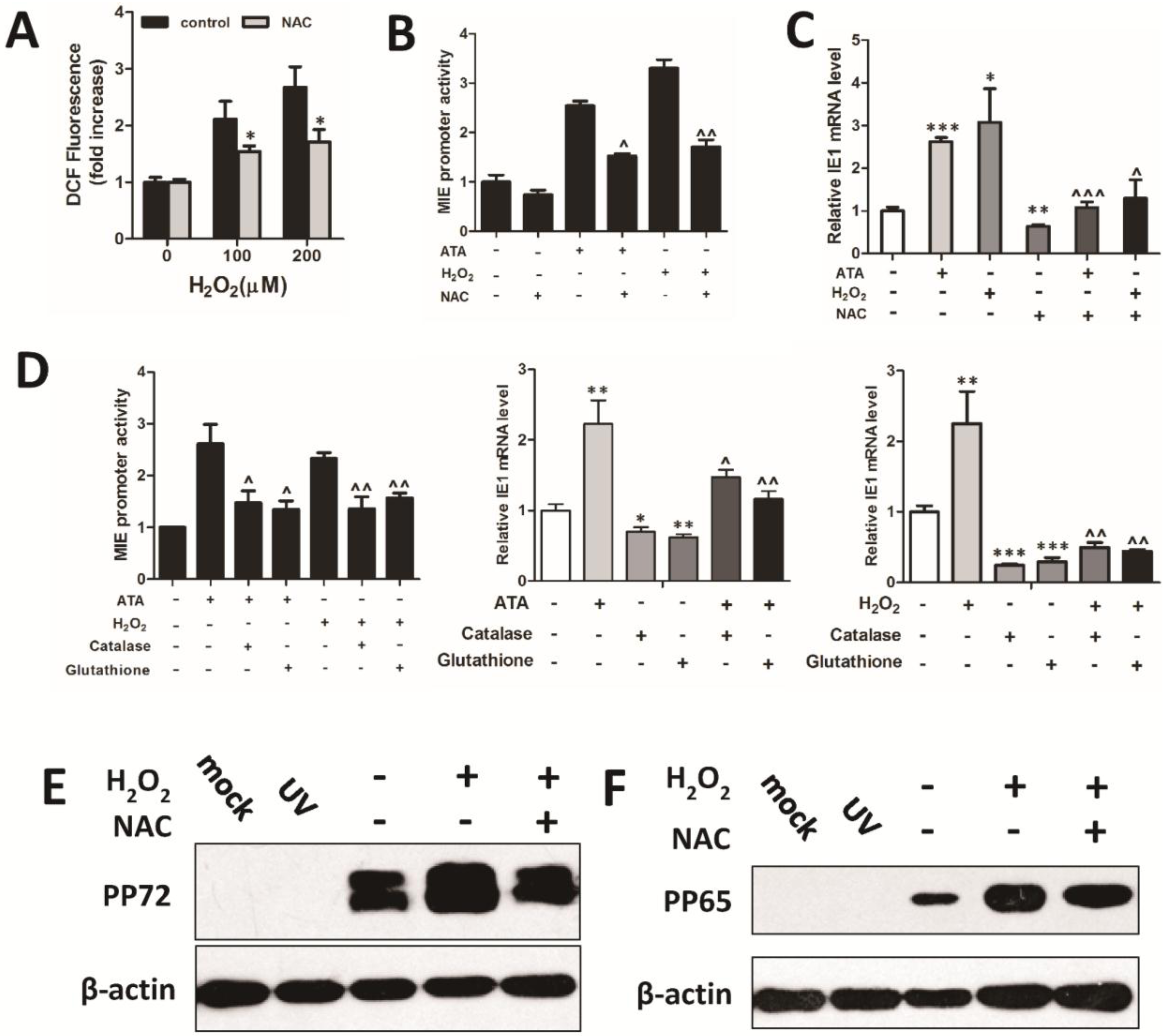
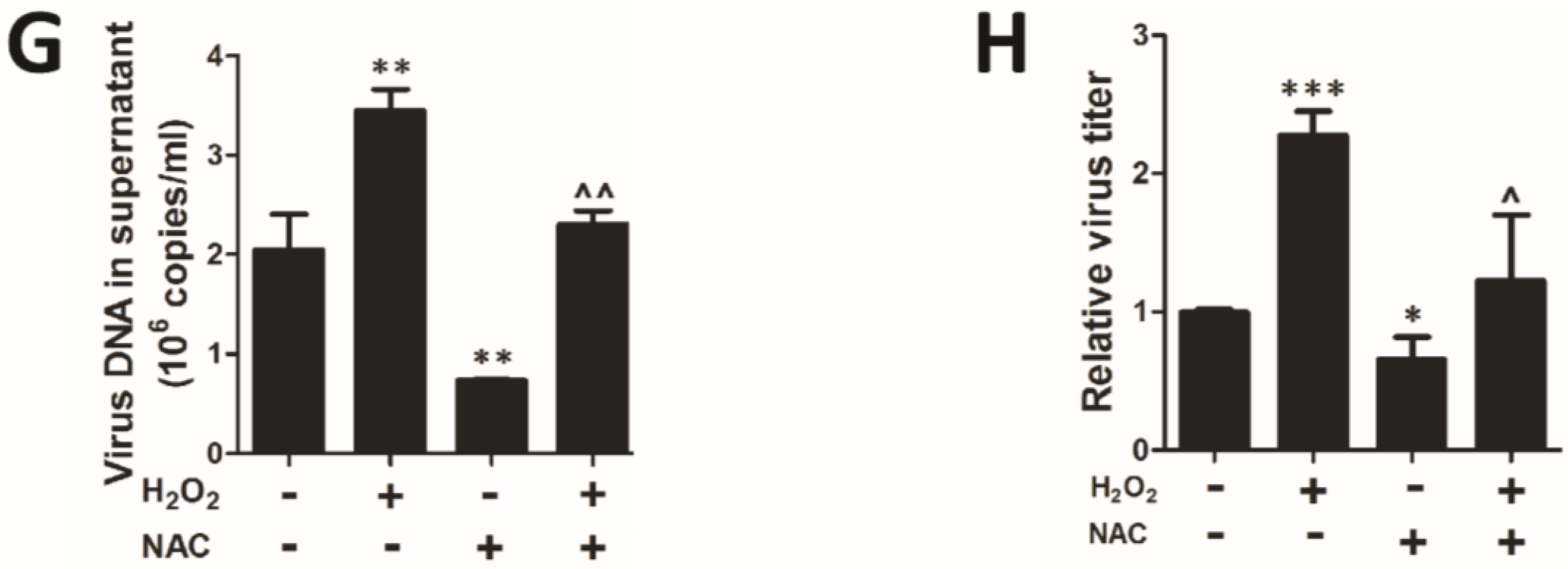
3.2. H2O2 Scavengers Inhibit H2O2-Upregulated HCMV Lytic Replication


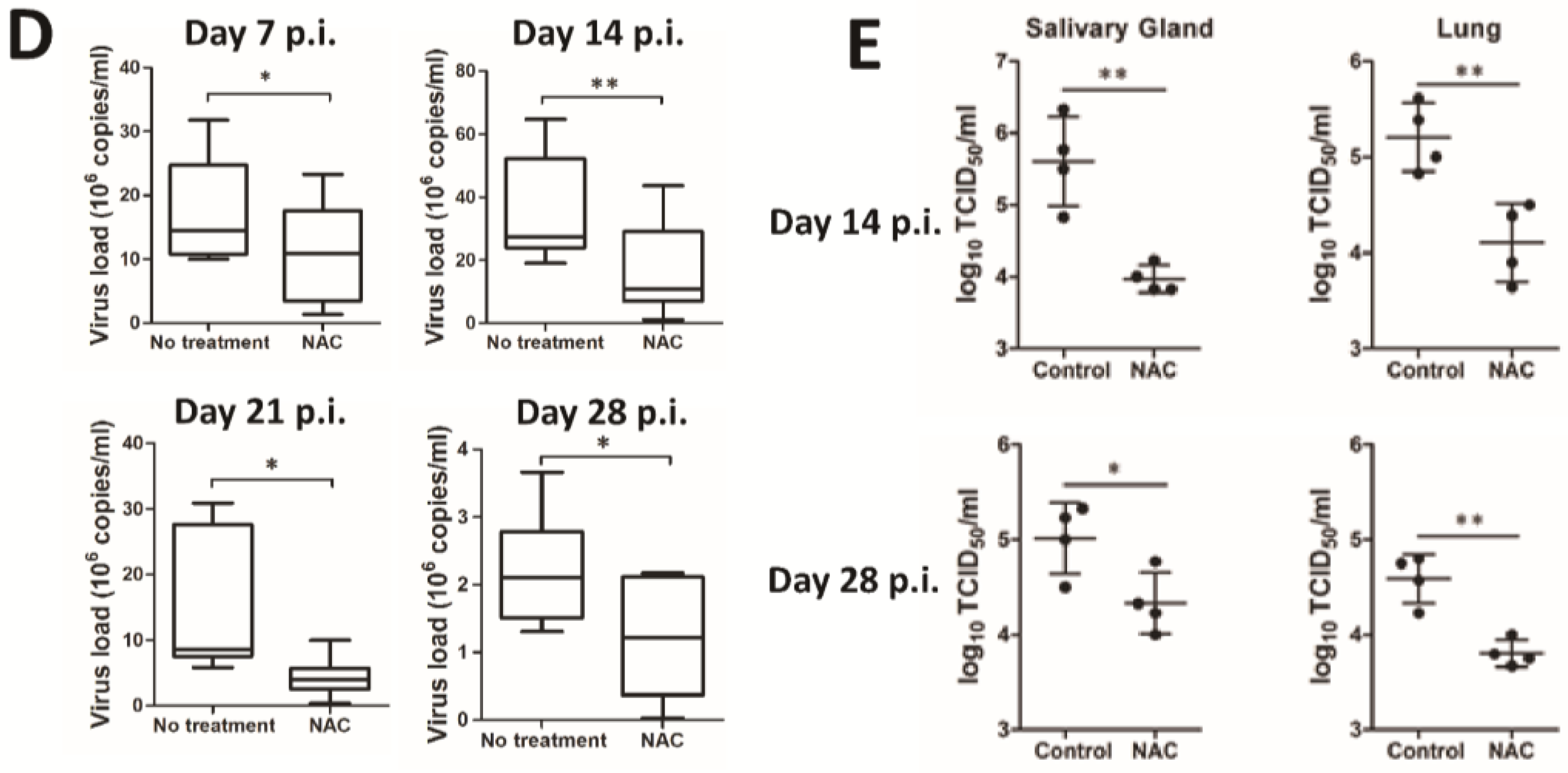
3.3. H2O2 Scavenger NAC Inhibits MCMV Lytic Replication in Vivo
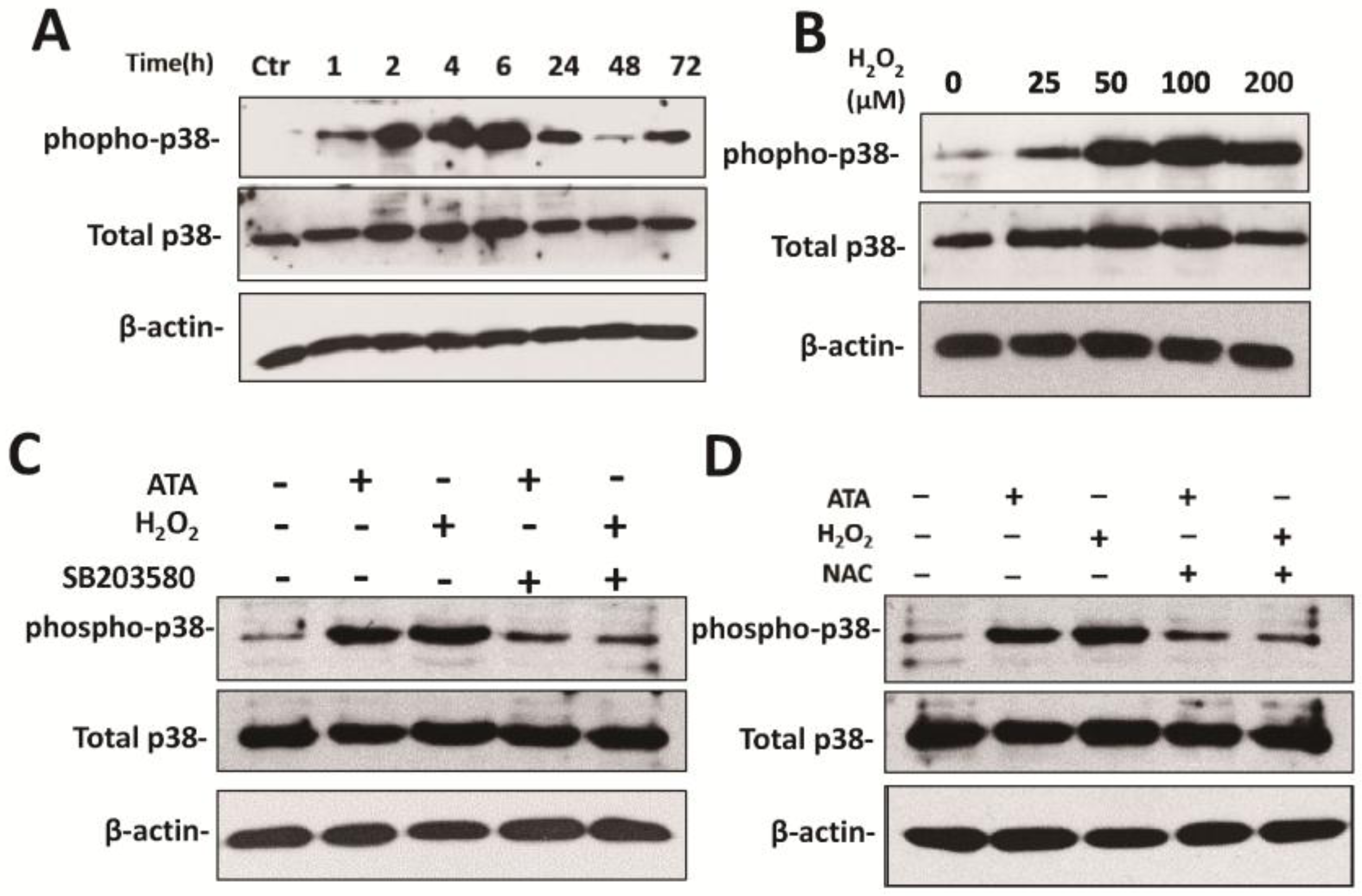
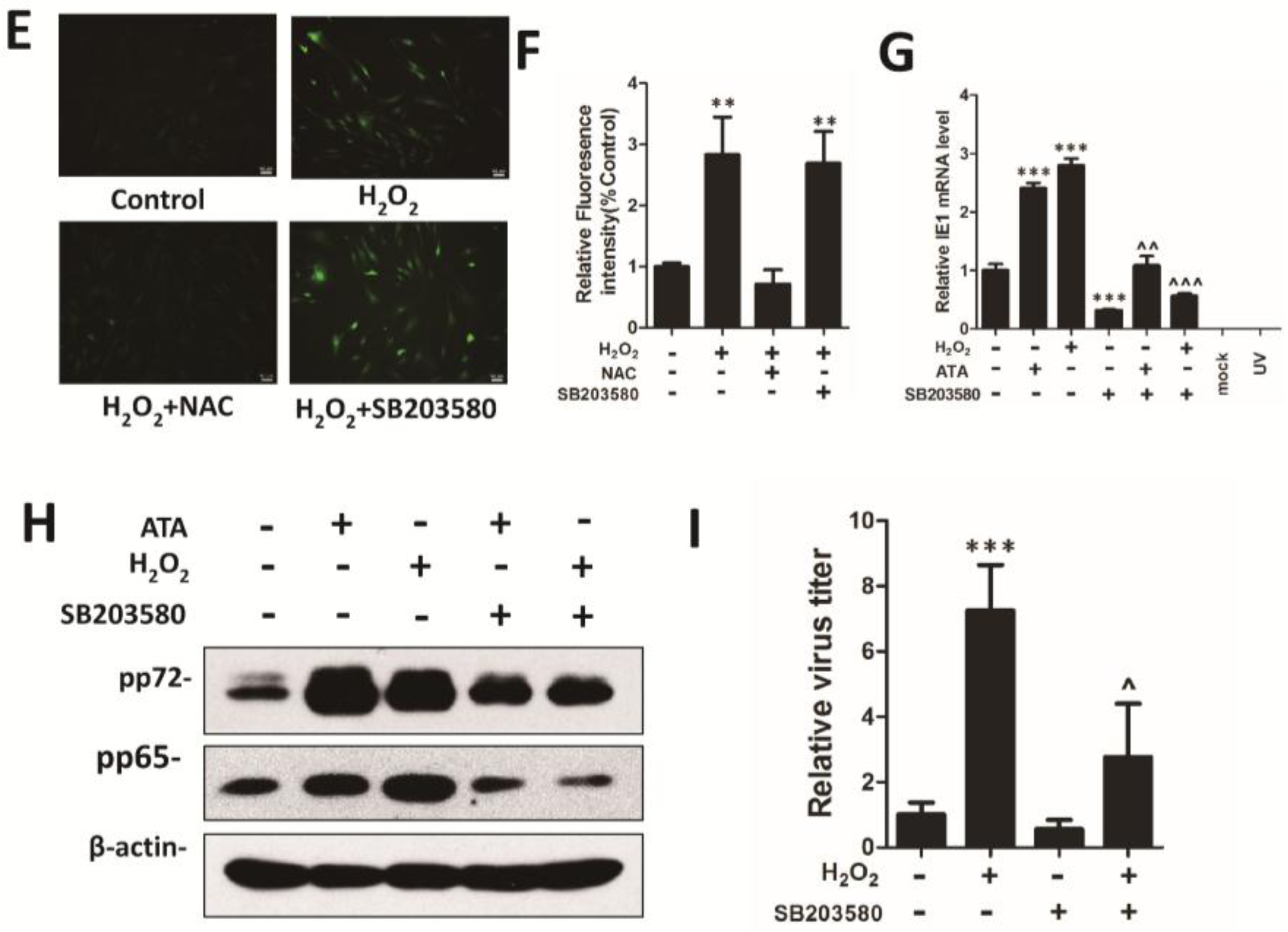
3.4. H2O2 Upregulates HCMV Replication by Activating the p38 MAPK Pathway




4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gerna, G.; Baldanti, F.; Revello, M.G. Pathogenesis of human cytomegalovirus infection and cellular targets. Hum. Immunol. 2004, 65, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Rubin, R.H. Impact of cytomegalovirus infection on organ transplant recipients. Rev. Infect. Dis. 1990, 12 (Suppl. 7), S754–S766. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Snydman, D.R.; Rubin, R.H.; Ho, M.; Pescovitz, M.; Martin, M.; Paya, C.V. Cytomegalovirus prophylaxis in solid organ transplant recipients. Transplantation 1996, 61, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Castro-Malaspina, H.; Harris, R.E.; Gajewski, J.; Ramsay, N.; Collins, R.; Dharan, B.; King, R.; Deeg, H.J. Unrelated donor marrow transplantation for myelodysplastic syndromes: Outcome analysis in 510 transplants facilitated by the National Marrow Donor Program. Blood 2002, 99, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- Steininger, C.; Puchhammer-Stockl, E.; Popow-Kraupp, T. Cytomegalovirus disease in the era of highly active antiretroviral therapy (HAART). J. Clin. Virol. 2006, 37, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Weis, M.; Kledal, T.N.; Lin, K.Y.; Panchal, S.N.; Gao, S.Z.; Valantine, H.A.; Mocarski, E.S.; Cooke, J.P. Cytomegalovirus infection impairs the nitric oxide synthase pathway: Role of asymmetric dimethylarginine in transplant arteriosclerosis. Circulation 2004, 109, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, J.; Fenton, M.; Dewar, C.; Ellins, E.; Storry, C.; Cubitt, D.; Deanfield, J.; Klein, N.; Halcox, J.; Burch, M. Endothelial dysfunction and cytomegalovirus replication in pediatric heart transplantation. Circulation 2008, 117, 2657–2661. [Google Scholar] [CrossRef] [PubMed]
- Arasaratnam, R.J. Cytomegalovirus and cardiovascular disease--the importance of covariates. J. Infect. Dis. 2013, 208, 1349. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, A.; Bostrom, L.; Lagerstedt, U.; Magnusson, I.; Soderberg-Naucler, C.; Sundqvist, V.A. Evidence of active cytomegalovirus infection and increased production of IL-6 in tissue specimens obtained from patients with inflammatory bowel diseases. Inflamm. Bowel Dis. 2003, 9, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Compton, T. Inhibition of inflammatory interleukin-6 activity via extracellular signal-regulated kinase-mitogen-activated protein kinase signaling antagonizes human cytomegalovirus reactivation from dendritic cells. J. Virol. 2011, 85, 12750–12758. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Breidenstein, A.; Compton, T. Human cytomegalovirus activation of ERK and myeloid cell leukemia-1 protein correlates with survival of latently infected cells. Proc. Natl. Acad. Sci. USA 2012, 109, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Rodems, S.M.; Spector, D.H. Extracellular signal-regulated kinase activity is sustained early during human cytomegalovirus infection. J. Virol. 1998, 72, 9173–9180. [Google Scholar] [PubMed]
- Johnson, R.A.; Ma, X.L.; Yurochko, A.D.; Huang, E.S. The role of MKK1/2 kinase activity in human cytomegalovirus infection. J. Gen. Virol. 2001, 82, 493–497. [Google Scholar] [PubMed]
- Jassem, W.; Fuggle, S.V.; Rela, M.; Koo, D.D.; Heaton, N.D. The role of mitochondria in ischemia/reperfusion injury. Transplantation 2002, 73, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, K.; Sporniak-Tutak, K.; Bober, J.; Safranow, K.; Olszewska, M.; Jakubowska, K.; Domanski, L.; Golembiewska, E.; Kwiatkowska, E.; Laszczynska, M.; et al. Oxidative stress indices in rats under immunosuppression. Transplant. Proc. 2011, 43, 3939–3945. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, F.; Mestre, E.; Essig, M.; Sauvage, F.L.; Marquet, P.; Gastinel, L.N. Quantitative proteomic analysis of cyclosporine-induced toxicity in a human kidney cell line and comparison with tacrolimus. J. Proteomics 2011, 75, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B. Oxidative stress in HIV patients receiving antiretroviral therapy. Curr. HIV Res. 2014, 12, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Carnevale, R.; Pignatelli, P. Is there a clinical role for oxidative stress biomarkers in atherosclerotic diseases? Intern. Emerg. Med. 2014, 9, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Forstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.S.; Hong, M.Z.; Ren, J.L. Reactive oxygen species: A double-edged sword in oncogenesis. World J. Gastroenterol. 2009, 15, 1702–1707. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, J. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys. Acta 2010, 1799, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Aggeli, I.K.; Gaitanaki, C.; Beis, I. Involvement of JNKs and p38-MAPK/MSK1 pathways in H2O2-induced upregulation of heme oxygenase-1 mRNA in H9c2 cells. Cell. Signal. 2006, 18, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Kefaloyianni, E.; Gaitanaki, C.; Beis, I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell. Signal. 2006, 18, 2238–2251. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.Q.; Zhang, Y.Y.; Lv, L.P.; Zhou, X.P.; Yan, F.; Ma, P.; Xu, J.B. Human cytomegalovirus-encoded chemokine receptor homolog US28 stimulates the major immediate early gene promoter/enhancer via the induction of CREB. J. Recept. Signal Transduct. Res. 2009, 29, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Keyes, L.R.; Bego, M.G.; Soland, M.; St Jeor, S. Cyclophilin A is required for efficient human cytomegalovirus DNA replication and reactivation. J. Gen. Virol. 2012, 93, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Nigro, P.; Matoba, T.; O’Dell, M.R.; Cui, Z.; Shi, X.; Mohan, A.; Yan, C.; Abe, J.; Illig, K.A.; et al. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat. Med. 2009, 15, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Rawlinson, W.D.; Farrell, H.E.; Barrell, B.G. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996, 70, 8833–8849. [Google Scholar] [PubMed]
- Krmpotic, A.; Bubic, I.; Polic, B.; Lucin, P.; Jonjic, S. Pathogenesis of murine cytomegalovirus infection. Microbes Infect./Inst. Pasteur 2003, 5, 1263–1277. [Google Scholar] [CrossRef]
- Bak, M.J.; Jeong, W.S.; Kim, K.B. Detoxifying effect of fermented black ginseng on H2O2-induced oxidative stress in HepG2 cells. Int. J. Mol. Med. 2014, 34, 1516–1522. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Cinatl, J.; Gross, V.; Vogel, J.U.; Blaheta, R.A.; Freisleben, H.J.; Markus, B.H.; Doerr, H.W. Impact of oxidative stress on human cytomegalovirus replication and on cytokine-mediated stimulation of endothelial cells. Transplantation 1996, 61, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Speir, E.; Shibutani, T.; Yu, Z.X.; Ferrans, V.; Epstein, S.E. Role of reactive oxygen intermediates in cytomegalovirus gene expression and in the response of human smooth muscle cells to viral infection. Circ. Res. 1996, 79, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Feng, J.; Sun, R. Oxidative stress induces reactivation of Kaposi’s sarcoma-associated herpesvirus and death of primary effusion lymphoma cells. J. Virol. 2011, 85, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhou, F.; Bedolla, R.G.; Jones, T.; Lei, X.; Kang, T.; Guadalupe, M.; Gao, S.J. Reactive oxygen species hydrogen peroxide mediates Kaposi's sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 2011, 7, e1002054. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Chen, Y.; Seth, S.; Furukawa, S.; Compans, R.W.; Jones, D.P. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 2003, 34, 928–936. [Google Scholar] [CrossRef]
- Staal, F.J.; Roederer, M.; Herzenberg, L.A.; Herzenberg, L.A. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1990, 87, 9943–9947. [Google Scholar] [CrossRef] [PubMed]
- McGuire, K.A.; Barlan, A.U.; Griffin, T.M.; Wiethoff, C.M. Adenovirus type 5 rupture of lysosomes leads to cathepsin B-dependent mitochondrial stress and production of reactive oxygen species. J. Virol. 2011, 85, 10806–10813. [Google Scholar] [CrossRef] [PubMed]
- Barlan, A.U.; Griffin, T.M.; McGuire, K.A.; Wiethoff, C.M. Adenovirus membrane penetration activates the NLRP3 inflammasome. J. Virol. 2011, 85, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Tung, W.H.; Hsieh, H.L.; Lee, I.T.; Yang, C.M. Enterovirus 71 induces integrin beta1/EGFR-Rac1-dependent oxidative stress in SK-N-SH cells: Role of HO-1/CO in viral replication. J. Cell. Physiol. 2011, 226, 3316–3329. [Google Scholar] [CrossRef] [PubMed]
- Kavouras, J.H.; Prandovszky, E.; Valyi-Nagy, K.; Kovacs, S.K.; Tiwari, V.; Kovacs, M.; Shukla, D.; Valyi-Nagy, T. Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse P19N neural cell cultures. J. Neurovirol. 2007, 13, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Aubert, M.; Chen, Z.; Lang, R.; Dang, C.H.; Fowler, C.; Sloan, D.D.; Jerome, K.R. The antiapoptotic herpes simplex virus glycoprotein J localizes to multiple cellular organelles and induces reactive oxygen species formation. J. Virol. 2008, 82, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, Y.H.; Utama, B.; Wang, J.; LeMaire, S.A.; Coselli, J.S.; Vercellotti, G.M.; Wang, X.L. HCMV infection attenuates hydrogen peroxide induced endothelial apoptosis-involvement of ERK pathway. FEBS Lett. 2006, 580, 2779–2787. [Google Scholar] [CrossRef] [PubMed]
- Tilton, C.; Clippinger, A.J.; Maguire, T.; Alwine, J.C. Human cytomegalovirus induces multiple means to combat reactive oxygen species. J. Virol. 2011, 85, 12585–12593. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, J.; Sissons, P. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J. Physiol. Pharmacol. 2013, 64, 409–421. [Google Scholar]
- Chen, K.; Vita, J.A.; Berk, B.C.; Keaney, J.F., Jr. c-Jun N-terminal kinase activation by hydrogen peroxide in endothelial cells involves SRC-dependent epidermal growth factor receptor transactivation. J. Biol. Chem. 2001, 276, 16045–16050. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, J.; Deng, J.; Lv, L.; Kang, Q.; Ma, P.; Yan, F.; Song, X.; Gao, B.; Zhang, Y.; Xu, J. Hydrogen Peroxide Induce Human Cytomegalovirus Replication through the Activation of p38-MAPK Signaling Pathway. Viruses 2015, 7, 2816-2833. https://doi.org/10.3390/v7062748
Xiao J, Deng J, Lv L, Kang Q, Ma P, Yan F, Song X, Gao B, Zhang Y, Xu J. Hydrogen Peroxide Induce Human Cytomegalovirus Replication through the Activation of p38-MAPK Signaling Pathway. Viruses. 2015; 7(6):2816-2833. https://doi.org/10.3390/v7062748
Chicago/Turabian StyleXiao, Jun, Jiang Deng, Liping Lv, Qiong Kang, Ping Ma, Fan Yan, Xin Song, Bo Gao, Yanyu Zhang, and Jinbo Xu. 2015. "Hydrogen Peroxide Induce Human Cytomegalovirus Replication through the Activation of p38-MAPK Signaling Pathway" Viruses 7, no. 6: 2816-2833. https://doi.org/10.3390/v7062748
APA StyleXiao, J., Deng, J., Lv, L., Kang, Q., Ma, P., Yan, F., Song, X., Gao, B., Zhang, Y., & Xu, J. (2015). Hydrogen Peroxide Induce Human Cytomegalovirus Replication through the Activation of p38-MAPK Signaling Pathway. Viruses, 7(6), 2816-2833. https://doi.org/10.3390/v7062748