
APOBEC3 Interference during Replication of Viral Genomes

Abstract
:1. APOBEC3s Edit Single-Stranded DNA
2. APOBEC3 Edition during Viral Replication Cycles
2.1. Retroviruses
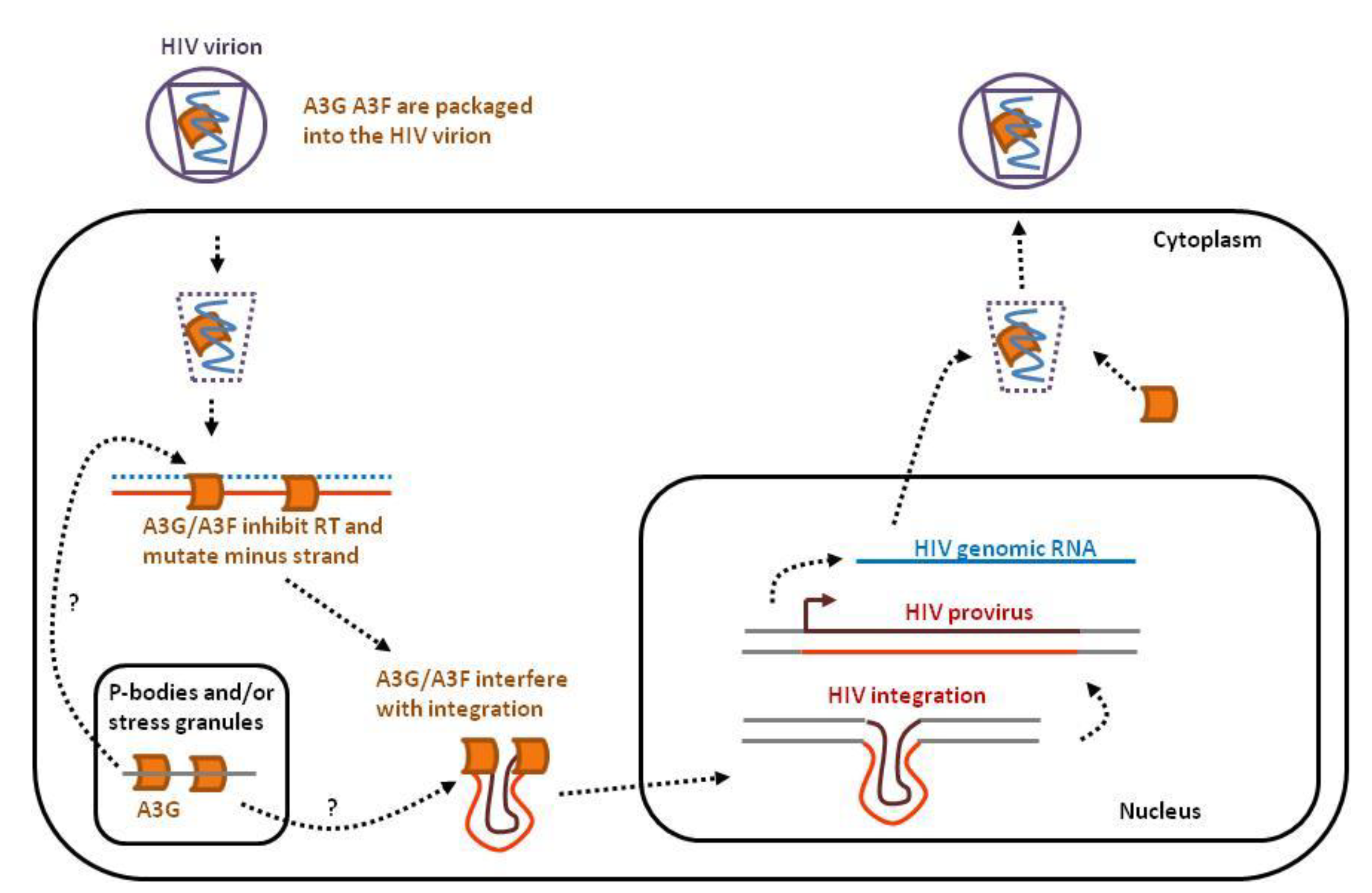
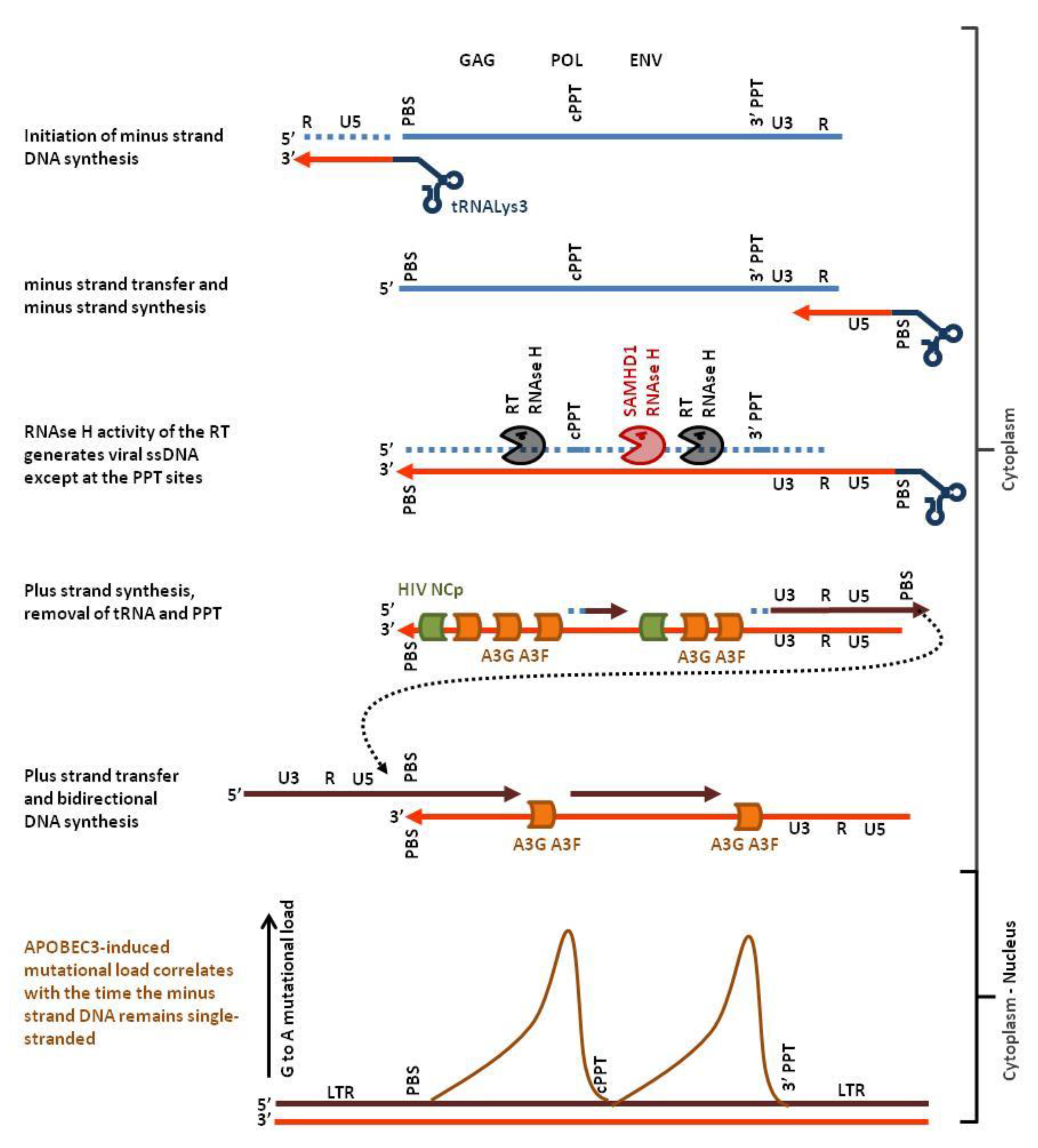
2.1.1. HIV-1


2.1.2. HTLV-1
2.1.3. HERVs
2.1.4. Simian Foamy Virus
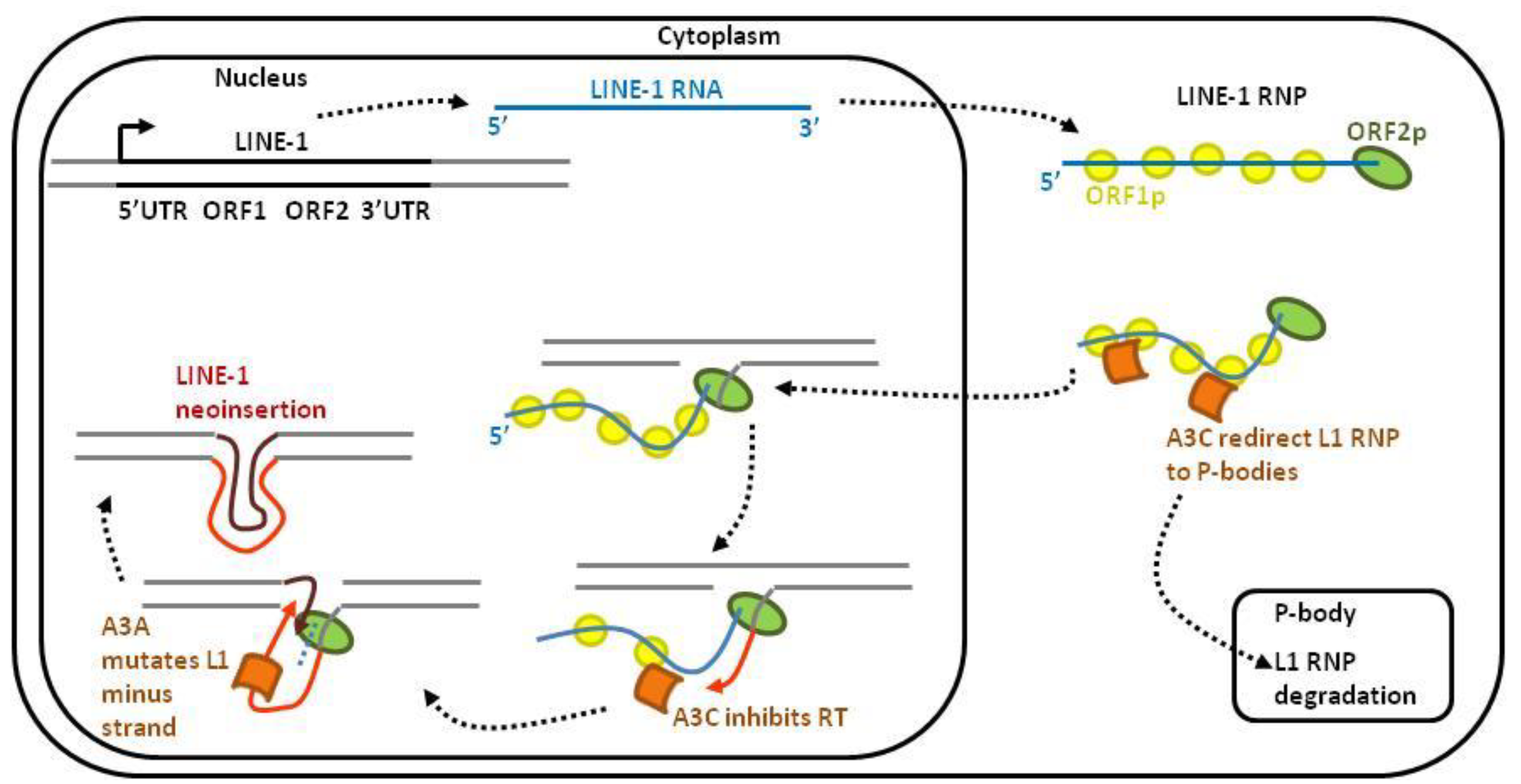
2.2. Retroelements

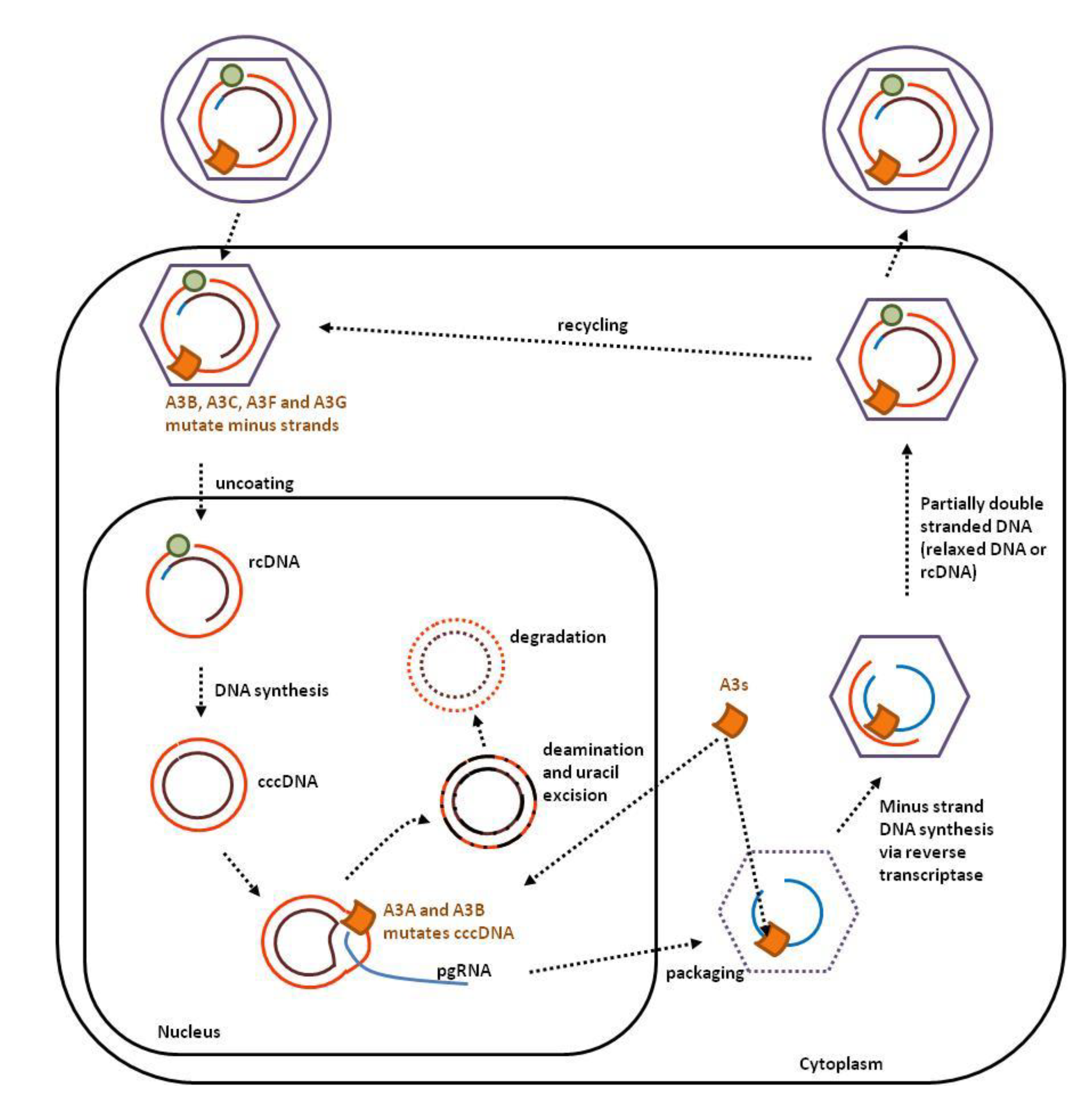
2.3. Hepadnaviruses


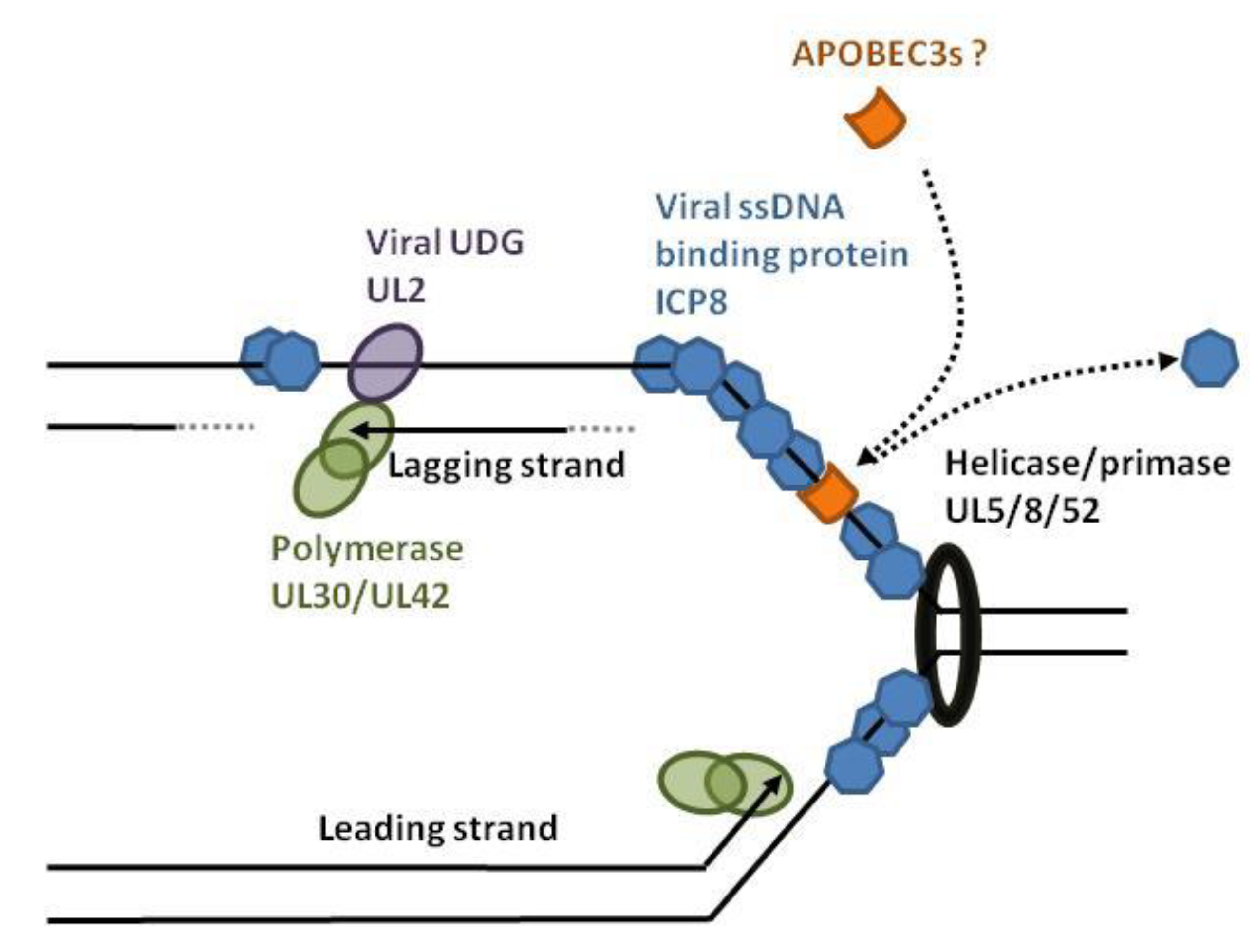
2.4. Herpesviruses

2.5. Papillomavirus
2.6. TT Virus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub-cellular localization | Substrate edited | Retro viruses | Retro elements | Hepadna viruses | Herpes viruses | ||||
|---|---|---|---|---|---|---|---|---|---|
| HIV-1 | HTLV-1 | HERVs | SFV | ||||||
| A3A | cell wide | single stranded DNA, RNA * | + | + | + | ||||
| A3B | nuclear | single stranded DNA | + | ||||||
| A3C | cell wide | single stranded DNA | + | + | + | ||||
| A3DE | cytoplasmic | single stranded DNA | + | ||||||
| A3F | cytoplasmic | single stranded DNA | + | + | + | ||||
| A3G | cytoplasmic | single stranded DNA | + | + | + | + | + | ||
| A3H | cell wide | single stranded DNA | + | + | + | ||||
3. Therapeutic Strategies by Perturbation of the Viral Mutation Rate

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smith, H.C.; Bennett, R.P.; Kizilyer, A.; McDougall, W.M.; Prohaska, K.M. Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol. 2012, 23, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Refsland, E.W.; Harris, R.S. The APOBEC3 family of retroelement restriction factors. Curr. Top. Microbiol. Immunol. 2013, 371, 1–27. [Google Scholar] [PubMed]
- Vieira, V.C.; Soares, M.A. The role of cytidine deaminases on innate immune responses against human viral infections. Biomed. Res. Int. 2013, 2013, e683095. [Google Scholar] [CrossRef] [PubMed]
- LaRue, R.S.; Andresdottir, V.; Blanchard, Y.; Conticello, S.G.; Derse, D.; Emerman, M.; Greene, W.C.; Jonsson, S.R.; Landau, N.R.; Lochelt, M.; et al. Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 2009, 83, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Munk, C.; Willemsen, A.; Bravo, I.G. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol. Biol. 2012, 12, e71. [Google Scholar] [CrossRef] [PubMed]
- Mehta, H.V.; Jones, P.H.; Weiss, J.P.; Okeoma, C.M. IFN-alpha and lipopolysaccharide upregulate APOBEC3 mRNA through different signaling pathways. J. Immunol. 2012, 189, 4088–4103. [Google Scholar] [CrossRef] [PubMed]
- Refsland, E.W.; Stenglein, M.D.; Shindo, K.; Albin, J.S.; Brown, W.L.; Harris, R.S. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction. Nucleic Acids Res. 2010, 38, 4274–4284. [Google Scholar] [CrossRef] [PubMed]
- Koning, F.A.; Newman, E.N.; Kim, E.Y.; Kunstman, K.J.; Wolinsky, S.M.; Malim, M.H. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009, 83, 9474–9485. [Google Scholar] [CrossRef] [PubMed]
- Lackey, L.; Law, E.K.; Brown, W.L.; Harris, R.S. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle 2013, 12, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Saetrom, P.; Aas, P.A.; Pettersen, H.S.; Kavli, B.; Slupphaug, G. Error-free versus mutagenic processing of genomic uracil—Relevance to cancer. DNA Repair 2014, 19, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Weil, A.F.; Ghosh, D.; Zhou, Y.; Seiple, L.; McMahon, M.A.; Spivak, A.M.; Siliciano, R.F.; Stivers, J.T. Uracil DNA glycosylase initiates degradation of HIV-1 cDNA containing misincorporated dUTP and prevents viral integration. Proc. Natl. Acad Sci. USA 2013, 110, E448–E457. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Huthoff, H.; Autore, F.; Gallois-Montbrun, S.; Fraternali, F.; Malim, M.H. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 2009, 5, e1000330. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Cen, S.; Niu, M.; Saadatmand, J.; Kleiman, L. Inhibition of tRNA(3)(Lys)—Primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 2006, 80, 11710–11722. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Hope, T.J. APOBEC3G restricts early HIV-1 replication in the cytoplasm of target cells. Virology 2008, 375, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ao, Z.; Chen, L.; Kobinger, G.; Peng, J.; Yao, X. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J. Virol. 2012, 86, 3777–3786. [Google Scholar] [CrossRef] [PubMed]
- Gillick, K.; Pollpeter, D.; Phalora, P.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J. Virol. 2013, 87, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Gummuluru, S.; Hu, J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 2007, 81, 4465–4472. [Google Scholar] [CrossRef] [PubMed]
- Narvaiza, I.; Linfesty, D.C.; Greener, B.N.; Hakata, Y.; Pintel, D.J.; Logue, E.; Landau, N.R.; Weitzman, M.D. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 2009, 5, e1000439. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.V.; Klawitter, S.; Held, U.; Berger, A.; Vasudevan, A.A.; Bock, A.; Hofmann, H.; Hanschmann, K.M.; Trosemeier, J.H.; Flory, E.; et al. Human LINE-1 restriction by APOBEC3C is deaminase independent and mediated by an ORF1p interaction that affects LINE reverse transcriptase activity. Nucleic Acids Res. 2014, 42, 396–416. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Wang, T.; Liu, B.; Tian, C.; Xiao, Z.; Kappes, J.; Yu, X.F. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 2007, 81, 7238–7248. [Google Scholar] [CrossRef] [PubMed]
- Mbisa, J.L.; Barr, R.; Thomas, J.A.; Vandegraaff, N.; Dorweiler, I.J.; Svarovskaia, E.S.; Brown, W.L.; Mansky, L.M.; Gorelick, R.J.; Harris, R.S.; et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 2007, 81, 7099–7110. [Google Scholar] [CrossRef] [PubMed]
- Vetter, M.L.; D’Aquila, R.T. Cytoplasmic APOBEC3G restricts incoming Vif-positive human immunodeficiency virus type 1 and increases two-long terminal repeat circle formation in activated T-helper-subtype cells. J. Virol. 2009, 83, 8646–8654. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Liddament, M.T.; Brown, W.L.; Schumacher, A.J.; Harris, R.S. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 2004, 14, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, H.L.; Doehle, B.P.; Bogerd, H.P.; Cullen, B.R. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004, 23, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Witkowska, H.E.; Hall, S.C.; Santiago, M.; Soros, V.B.; Esnault, C.; Heidmann, T.; Greene, W.C. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. USA 2006, 103, 15588–15593. [Google Scholar] [CrossRef] [PubMed]
- Gallois-Montbrun, S.; Kramer, B.; Swanson, C.M.; Byers, H.; Lynham, S.; Ward, M.; Malim, M.H. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 2007, 81, 2165–2178. [Google Scholar] [CrossRef] [PubMed]
- Wichroski, M.J.; Robb, G.B.; Rana, T.M. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2006, 2, e41. [Google Scholar] [CrossRef] [PubMed]
- Phalora, P.K.; Sherer, N.M.; Wolinsky, S.M.; Swanson, C.M.; Malim, M.H. HIV-1 replication and APOBEC3 antiviral activity are not regulated by P bodies. J. Virol. 2012, 86, 11712–11724. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Koizumi, Y.; Takeuchi, J.S.; Misawa, N.; Kimura, Y.; Morita, S.; Aihara, K.; Koyanagi, Y.; Iwami, S.; Sato, K. Quantification of deaminase activity-dependent and -independent restriction of HIV-1 replication mediated by APOBEC3F and APOBEC3G through experimental-mathematical investigation. J. Virol. 2014, 88, 5881–5887. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Rusniok, C.; Vartanian, J.P.; Wain-Hobson, S. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 2006, 34, 4677–4684. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Corona, A.; Tramontano, E. HIV-1 Reverse Transcriptase Still Remains a New Drug Target: Structure, Function, Classical Inhibitors, and New Inhibitors with Innovative Mechanisms of Actions. Mol. Biol. Int. 2012, 2012, e586401. [Google Scholar] [CrossRef] [PubMed]
- Mariani, R.; Chen, D.; Schrofelbauer, B.; Navarro, F.; Konig, R.; Bollman, B.; Munk, C.; Nymark-McMahon, H.; Landau, N.R. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 2003, 114, 21–31. [Google Scholar] [CrossRef]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 2012, 481, 371–375. [Google Scholar]
- Zhao, K.; Du, J.; Rui, Y.; Zheng, W.; Kang, J.; Hou, J.; Wang, K.; Zhang, W.; Simon, V.A.; Yu, X.F. Evolutionarily conserved pressure for the existence of distinct G2/M cell cycle arrest and A3H inactivation functions in HIV-1 Vif. Cell Cycle 2015, 14, 838–847. [Google Scholar] [PubMed]
- Stavrou, S.; Nitta, T.; Kotla, S.; Ha, D.; Nagashima, K.; Rein, A.R.; Fan, H.; Ross, S.R. Murine leukemia virus glycosylated Gag blocks apolipoprotein B editing complex 3 and cytosolic sensor access to the reverse transcription complex. Proc. Natl. Acad. Sci. USA 2013, 110, 9078–9083. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.; Hercik, K.; Byeon, I.J.; Ahn, J.; Hill, S.; Hinchee-Rodriguez, K.; Singer, D.; Byeon, C.H.; Charlton, L.M.; Nam, G.; et al. Structural determinants of human APOBEC3A enzymatic and nucleic acid binding properties. Nucleic Acids Res. 2014, 42, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Beloglazova, N.; Flick, R.; Tchigvintsev, A.; Brown, G.; Popovic, A.; Nocek, B.; Yakunin, A.F. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 2013, 288, 8101–8110. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, J.; Choi, J.; Oh, C.; Kim, S.; Seo, M.; Kim, S.Y.; Seo, D.; Kim, J.; White, T.E.; Brandariz-Nunez, A.; et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 2014, 20, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Mahieux, R.; Suspene, R.; Delebecque, F.; Henry, M.; Schwartz, O.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J. Gen. Virol. 2005, 86, 2489–2494. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ma, G.; Nosaka, K.; Tanabe, J.; Satou, Y.; Koito, A.; Wain-Hobson, S.; Vartanian, J.P.; Matsuoka, M. APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J. Virol. 2010, 84, 7278–7287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavrois, M.; Wain-Hobson, S.; Gessain, A.; Plumelle, Y.; Wattel, E. Adult T-cell leukemia/lymphoma on a background of clonally expanding human T-cell leukemia virus type-1-positive cells. Blood 1996, 88, 4646–4650. [Google Scholar] [PubMed]
- Etoh, K.; Tamiya, S.; Yamaguchi, K.; Okayama, A.; Tsubouchi, H.; Ideta, T.; Mueller, N.; Takatsuki, K.; Matsuoka, M. Persistent clonal proliferation of human T-lymphotropic virus type I-infected cells in vivo. Cancer Res. 1997, 57, 4862–4867. [Google Scholar] [PubMed]
- Mortreux, F.; Gabet, A.S.; Wattel, E. Molecular and cellular aspects of HTLV-1 associated leukemogenesis in vivo. Leukemia 2003, 17, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.A.; Malani, N.; Melamed, A.; Gormley, N.; Carter, R.; Bentley, D.; Berry, C.; Bushman, F.D.; Taylor, G.P.; Bangham, C.R. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood 2011, 117, 3113–3122. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Konig, R.; Pillai, S.; Chiles, K.; Kearney, M.; Palmer, S.; Richman, D.; Coffin, J.M.; Landau, N.R. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004, 11, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Derse, D.; Hill, S.A.; Princler, G.; Lloyd, P.; Heidecker, G. Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. Proc. Natl. Acad. Sci. USA 2007, 104, 2915–2920. [Google Scholar] [CrossRef] [PubMed]
- Medstrand, P.; van de Lagemaat, L.N.; Dunn, C.A.; Landry, J.R.; Svenback, D.; Mager, D.L. Impact of transposable elements on the evolution of mammalian gene regulation. Cytogenet. Genome Res. 2005, 110, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Stoye, J.P. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat. Rev. Microbiol. 2012, 10, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Malim, M.H.; Bieniasz, P.D. Hypermutation of an ancient human retrovirus by APOBEC3G. J. Virol. 2008, 82, 8762–8770. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Priet, S.; Ribet, D.; Heidmann, O.; Heidmann, T. Restriction by APOBEC3 proteins of endogenous retroviruses with an extracellular life cycle: Ex vivo effects and in vivo “traces” on the murine IAPE and human HERV-K elements. Retrovirology 2008, 5, e75. [Google Scholar] [CrossRef] [PubMed]
- Anwar, F.; Davenport, M.P.; Ebrahimi, D. Footprint of APOBEC3 on the genome of human retroelements. J. Virol. 2013, 87, 8195–8204. [Google Scholar] [CrossRef] [PubMed]
- Rua, R.; Gessain, A. Origin, evolution and innate immune control of simian foamy viruses in humans. Curr. Opin. Virol. 2015, 10, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Gartner, K.; Wiktorowicz, T.; Park, J.; Mergia, A.; Rethwilm, A.; Scheller, C. Accuracy estimation of foamy virus genome copying. Retrovirology 2009, 6, e32. [Google Scholar] [CrossRef] [PubMed]
- Delebecque, F.; Suspene, R.; Calattini, S.; Casartelli, N.; Saib, A.; Froment, A.; Wain-Hobson, S.; Gessain, A.; Vartanian, J.P.; Schwartz, O. Restriction of foamy viruses by APOBEC cytidine deaminases. J. Virol. 2006, 80, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Rua, R.; Betsem, E.; Gessain, A. Viral latency in blood and saliva of simian foamy virus-infected humans. PLoS ONE 2013, 8, e77072. [Google Scholar] [CrossRef] [PubMed]
- Matsen, F.A.T.; Small, C.T.; Soliven, K.; Engel, G.A.; Feeroz, M.M.; Wang, X.; Craig, K.L.; Hasan, M.K.; Emerman, M.; Linial, M.L.; et al. A novel bayesian method for detection of APOBEC3-mediated hypermutation and its application to zoonotic transmission of simian foamy viruses. PLoS Comput. Biol. 2014, 10, e1003493. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schafer, A.; McClure, M.O.; Cullen, B.R. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef] [PubMed]
- Lochelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rosler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, M.; Schmidt, S.; Marino, D.; Russell, R.A.; Stauch, B.; Hofmann, H.; Kopietz, F.; Kloke, B.P.; Zielonka, J.; Strover, H.; et al. Species-specific inhibition of APOBEC3C by the prototype foamy virus protein bet. J. Biol. Chem. 2009, 284, 5819–5826. [Google Scholar] [CrossRef] [PubMed]
- Jaguva Vasudevan, A.A.; Perkovic, M.; Bulliard, Y.; Cichutek, K.; Trono, D.; Haussinger, D.; Munk, C. Prototype foamy virus Bet impairs the dimerization and cytosolic solubility of human APOBEC3G. J. Virol. 2013, 87, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.R.; Narvaiza, I.; Planegger, R.A.; Weitzman, M.D.; Moran, J.V. APOBEC3A deaminates transiently exposed single-strand DNA during LINE-1 retrotransposition. eLife 2014, 3, e02008. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Guetard, D.; Henry, M.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 8321–8326. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Guetard, D.; Suspene, R.; Rusniok, C.; Wain-Hobson, S.; Vartanian, J.P. Genetic editing of HBV DNA by monodomain human APOBEC3 cytidine deaminases and the recombinant nature of APOBEC3G. PLoS ONE 2009, 4, e4277. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.P.; Henry, M.; Marchio, A.; Suspene, R.; Aynaud, M.M.; Guetard, D.; Cervantes-Gonzalez, M.; Battiston, C.; Mazzaferro, V.; Pineau, P.; et al. Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog. 2010, 6, e1000928. [Google Scholar] [CrossRef] [PubMed]
- Beggel, B.; Munk, C.; Daumer, M.; Hauck, K.; Haussinger, D.; Lengauer, T.; Erhardt, A. Full genome ultra-deep pyrosequencing associates G-to-A hypermutation of the hepatitis B virus genome with the natural progression of hepatitis B. J. Viral. Hepat. 2013, 20, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Aynaud, M.M.; Suspene, R.; Vidalain, P.O.; Mussil, B.; Guetard, D.; Tangy, F.; Wain-Hobson, S.; Vartanian, J.P. Human Tribbles 3 protects nuclear DNA from cytidine deamination by APOBEC3A. J. Biol. Chem. 2012, 287, 39182–39192. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Aynaud, M.M.; Koch, S.; Pasdeloup, D.; Labetoulle, M.; Gaertner, B.; Vartanian, J.P.; Meyerhans, A.; Wain-Hobson, S. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 2011, 85, 7594–7602. [Google Scholar] [CrossRef] [PubMed]
- Muylaert, I.; Tang, K.W.; Elias, P. Replication and recombination of herpes simplex virus DNA. J. Biol. Chem. 2011, 286, 15619–15624. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.K.; Coen, D.M. Herpes simplex viruses: Mechanisms of DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a013011. [Google Scholar] [CrossRef] [PubMed]
- Bogani, F.; Corredeira, I.; Fernandez, V.; Sattler, U.; Rutvisuttinunt, W.; Defais, M.; Boehmer, P.E. Association between the herpes simplex virus-1 DNA polymerase and uracil DNA glycosylase. J. Biol. Chem. 2010, 285, 27664–27672. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.P.; Guetard, D.; Henry, M.; Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wakae, K.; Kitamura, K.; Aoyama, S.; Liu, G.; Koura, M.; Monjurul, A.M.; Kukimoto, I.; Muramatsu, M. APOBEC3 deaminases induce hypermutation in human papillomavirus 16 DNA upon beta interferon stimulation. J. Virol. 2014, 88, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Melendy, T. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA. J. Virol. 2004, 78, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, M.; Noguchi, C.; Akiyama, R.; Matsushita, M.; Kunihiro, K.; Tanaka, S.; Abe, H.; Mitsui, F.; Kitamura, S.; Hatakeyama, T.; et al. G to A hypermutation of TT virus. Virus Res. 2010, 149, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Patnaik, S.K.; Thomas Taggart, R.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015, 6, 6881. [Google Scholar] [CrossRef] [PubMed]
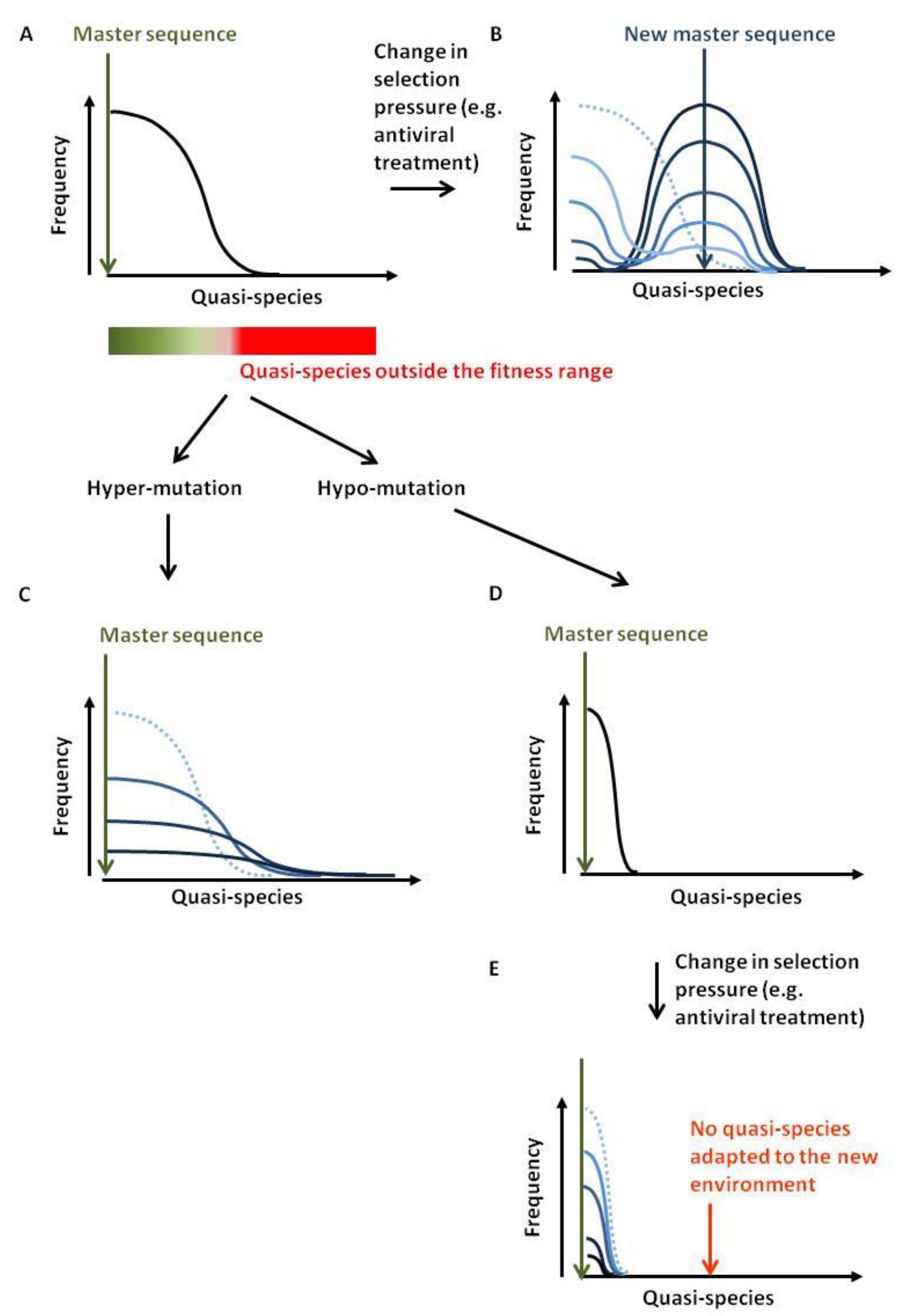
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, J.F.; Harris, R.S. Leveraging APOBEC3 proteins to alter the HIV mutation rate and combat AIDS. Future Virol. 2009, 4, 605. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S. Enhancing immunity to HIV through APOBEC. Nat. Biotechnol. 2008, 26, 1089–1090. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.J.; Nik-Zainal, S.; Wu, Y.L.; Stebbings, L.A.; Raine, K.; Campbell, P.J.; Rada, C.; Stratton, M.R.; Neuberger, M.S. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife 2013, 2, e00534. [Google Scholar] [CrossRef] [PubMed]
- Lada, A.G.; Dhar, A.; Boissy, R.J.; Hirano, M.; Rubel, A.A.; Rogozin, I.B.; Pavlov, Y.I. AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biol. Direct. 2012, 7, e47. [Google Scholar] [CrossRef] [PubMed]
- Lada, A.G.; Stepchenkova, E.I.; Waisertreiger, I.S.; Noskov, V.N.; Dhar, A.; Eudy, J.D.; Boissy, R.J.; Hirano, M.; Rogozin, I.B.; Pavlov, Y.I. Genome-wide mutation avalanches induced in diploid yeast cells by a base analog or an APOBEC deaminase. PLoS Genet. 2013, 9, e1003736. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Klimczak, L.J.; Kryukov, G.V.; Malc, E.; Mieczkowski, P.A.; et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 2012, 46, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Liu, D.; Lv, W.; Wang, X.; Wang, J.; Lv, M.; Huang, W.; Wu, J.; Zhang, H.; Jin, H.; et al. Small-molecule inhibition of human immunodeficiency virus type 1 replication by targeting the interaction between Vif and ElonginC. J. Virol. 2012, 86, 5497–5507. [Google Scholar] [CrossRef] [PubMed]
- Pery, E.; Sheehy, A.; Nebane, N.M.; Brazier, A.J.; Misra, V.; Rajendran, K.S.; Buhrlage, S.J.; Mankowski, M.K.; Rasmussen, L.; White, E.L.; et al. Identification of a Novel HIV-1 Inhibitor Targeting Vif-dependent Degradation of Human APOBEC3G. J. Biol. Chem. 2015, 290, 10504–10517. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Shandilya, S.M.; Carpenter, M.A.; Rathore, A.; Brown, W.L.; Perkins, A.L.; Harki, D.A.; Solberg, J.; Hook, D.J.; Pandey, K.K.; et al. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol. 2012, 7, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.E.; Li, M.; Harris, R.S.; Harki, D.A. Small-molecule APOBEC3G DNA cytosine deaminase inhibitors based on a 4-amino-1,2,4-triazole-3-thiol scaffold. ChemMedChem 2013, 8, 112–117. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willems, L.; Gillet, N.A. APOBEC3 Interference during Replication of Viral Genomes. Viruses 2015, 7, 2999-3018. https://doi.org/10.3390/v7062757
Willems L, Gillet NA. APOBEC3 Interference during Replication of Viral Genomes. Viruses. 2015; 7(6):2999-3018. https://doi.org/10.3390/v7062757
Chicago/Turabian StyleWillems, Luc, and Nicolas Albert Gillet. 2015. "APOBEC3 Interference during Replication of Viral Genomes" Viruses 7, no. 6: 2999-3018. https://doi.org/10.3390/v7062757
APA StyleWillems, L., & Gillet, N. A. (2015). APOBEC3 Interference during Replication of Viral Genomes. Viruses, 7(6), 2999-3018. https://doi.org/10.3390/v7062757