
Phylodynamics of H5N1 Highly Pathogenic Avian Influenza in Europe, 2005–2010: Potential for Molecular Surveillance of New Outbreaks

Abstract
:1. Introduction
2. Material and Methods
2.1. Sequence Data
2.2. Preliminary Phylogenetic Analysis
2.3. Divergence-Time Estimation
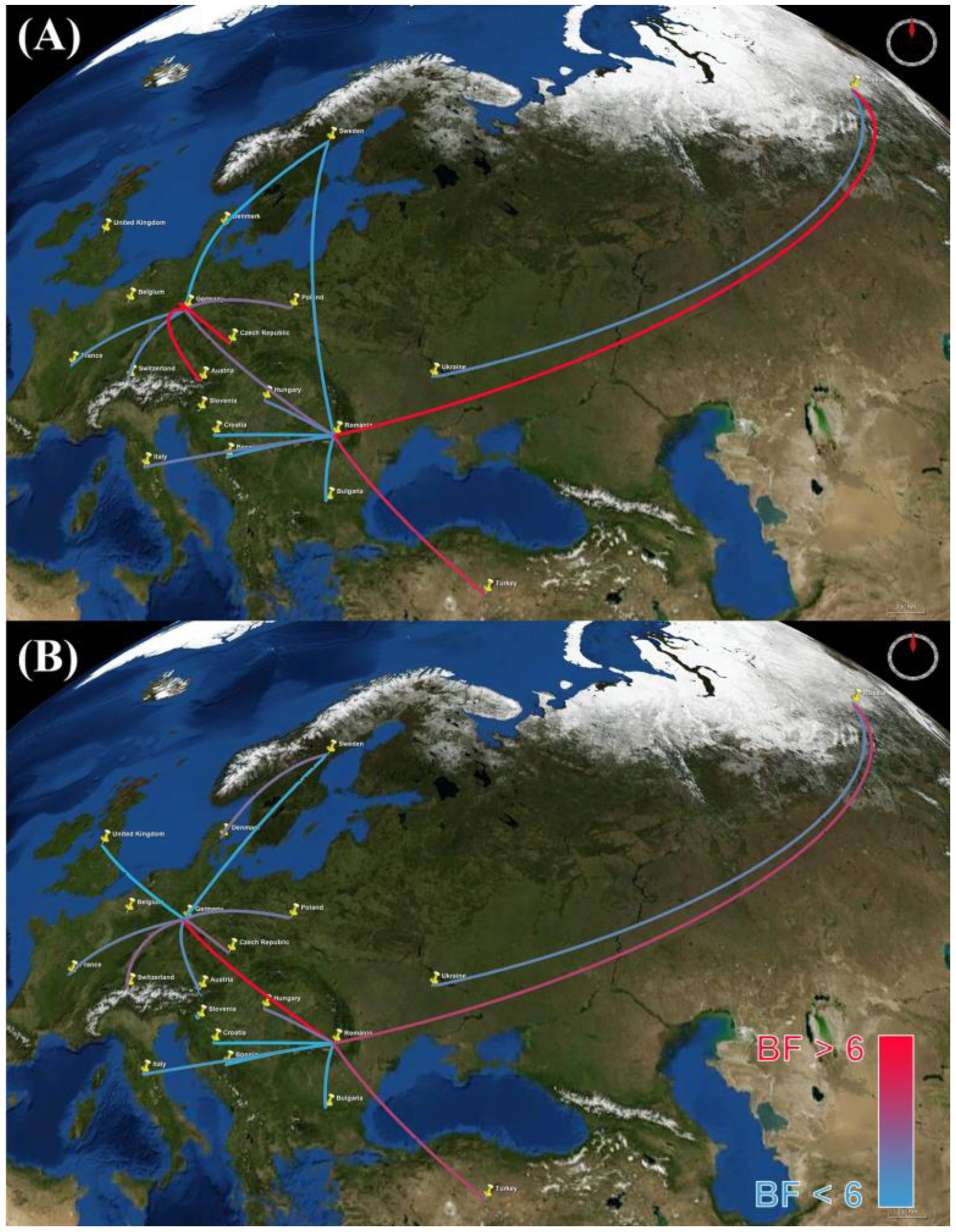
2.4. Estimation of Geographic History under the Discrete Phylodynamic Model
2.5. Exploring the Evolution of H5N1 HPAIV Host Infection
2.6. Assessing Uncertainty in Discrete-Trait Mappings and Association Statistics
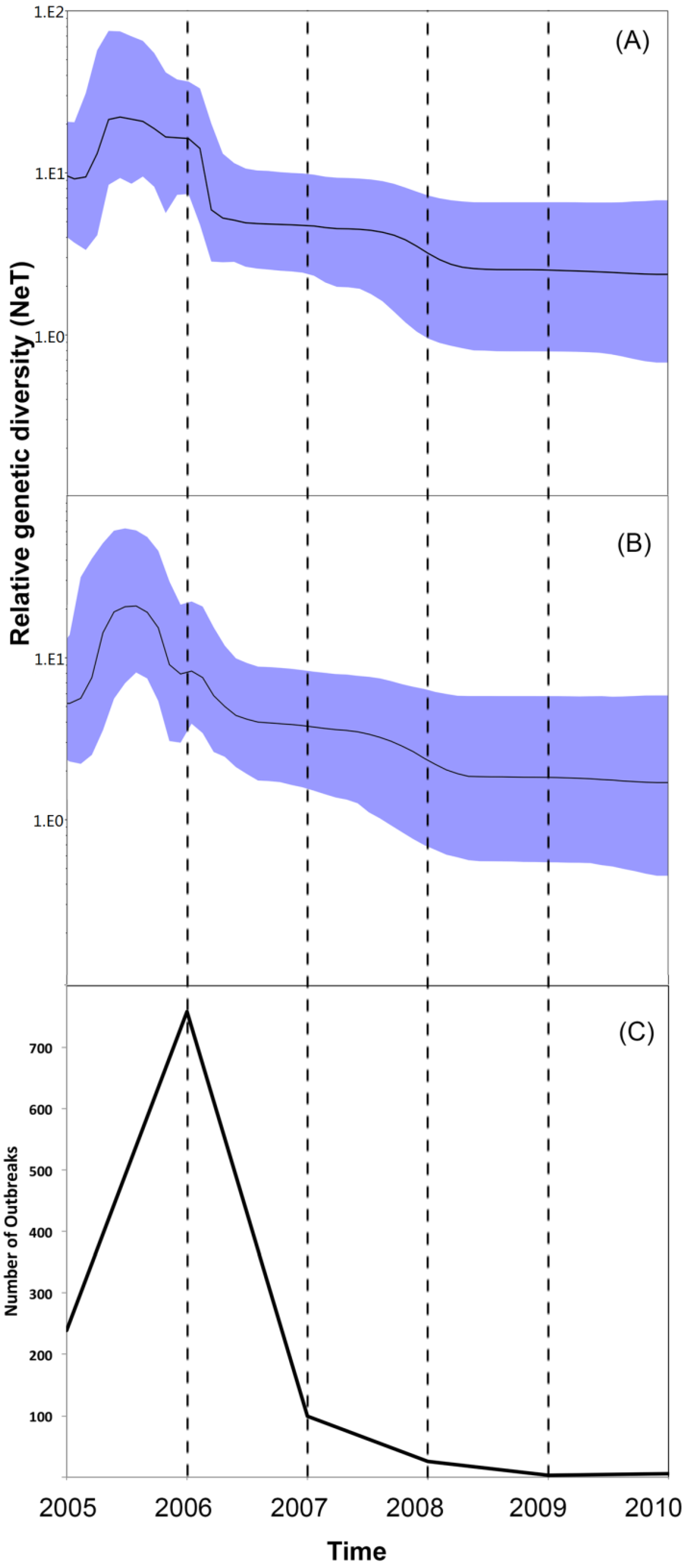
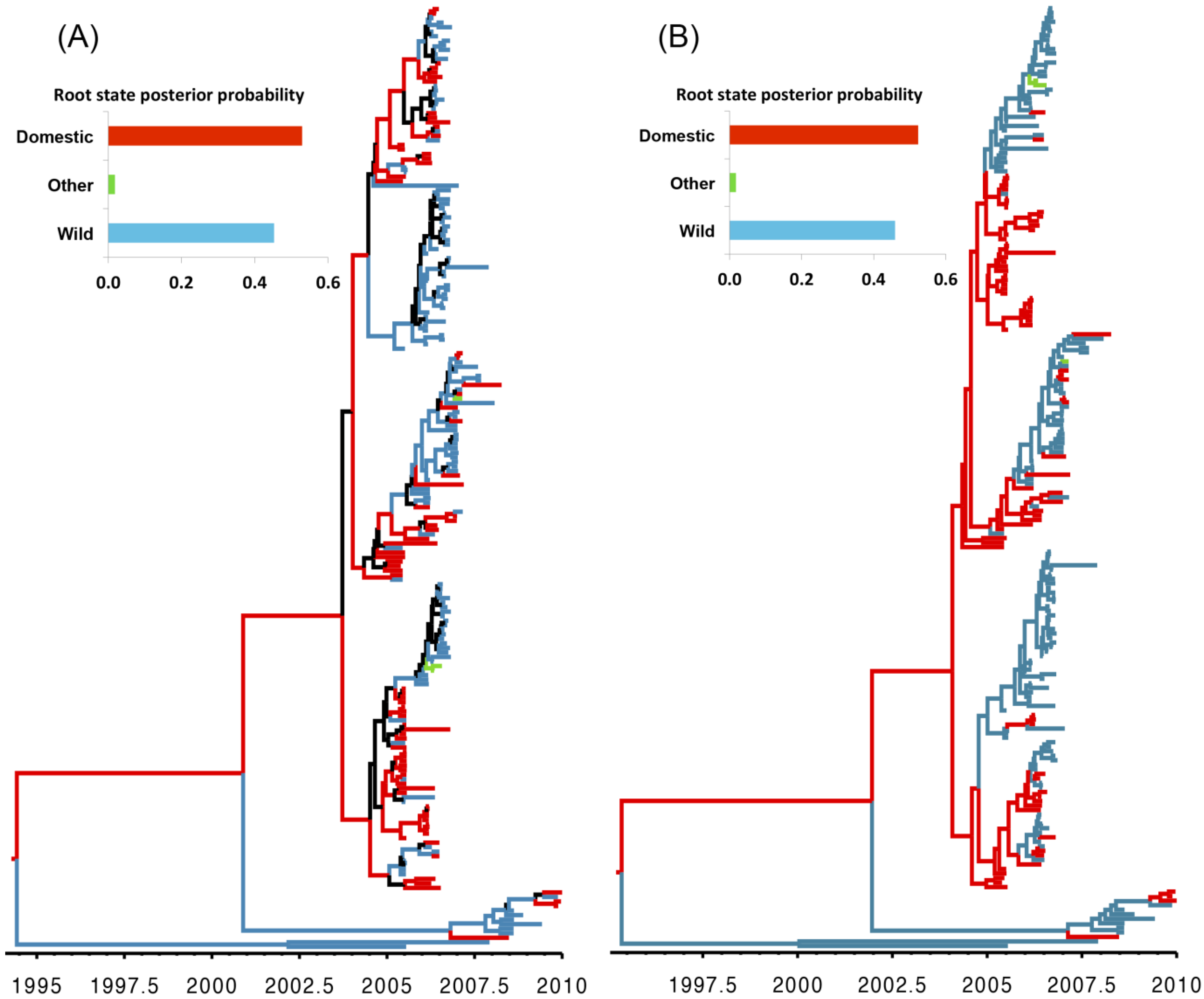
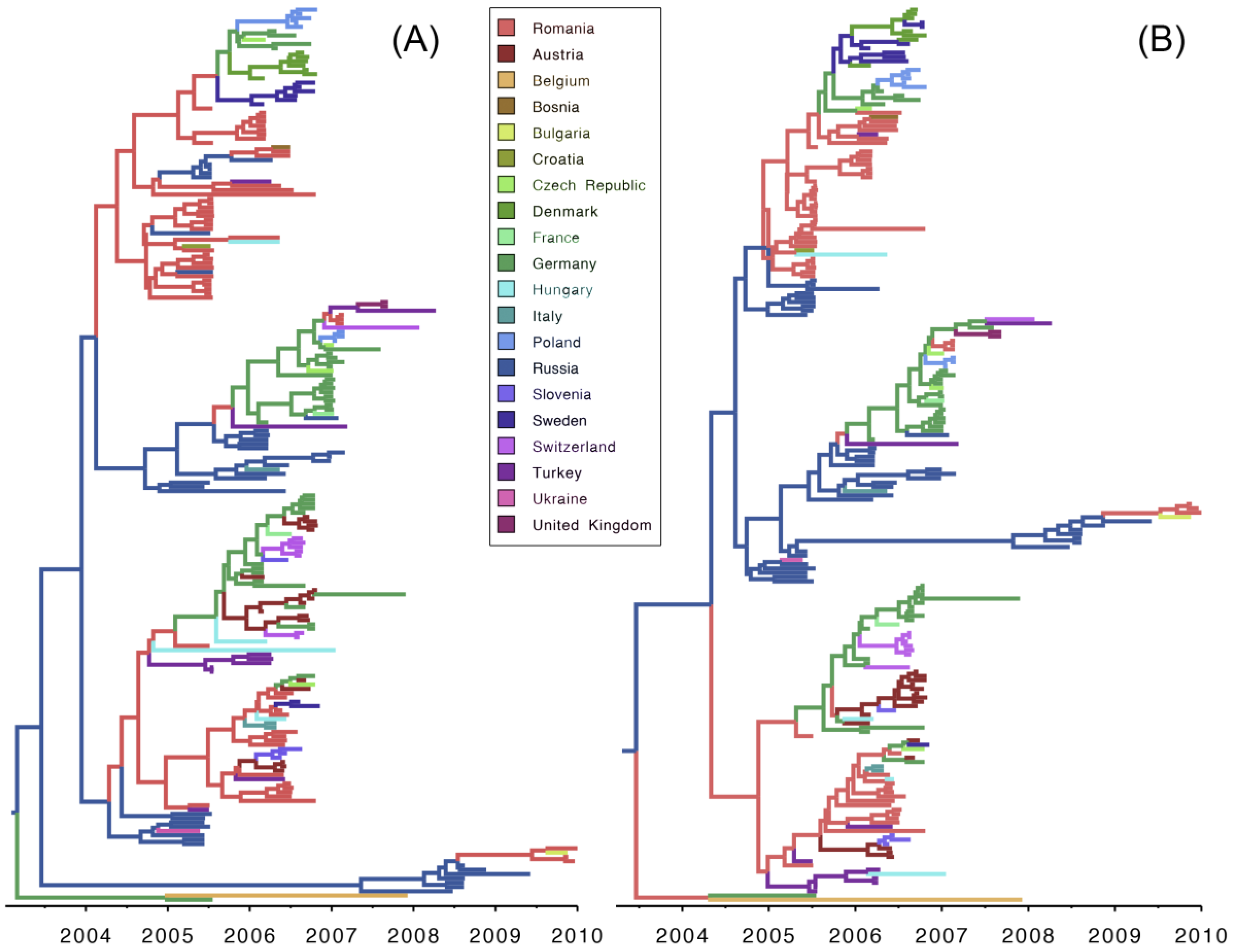
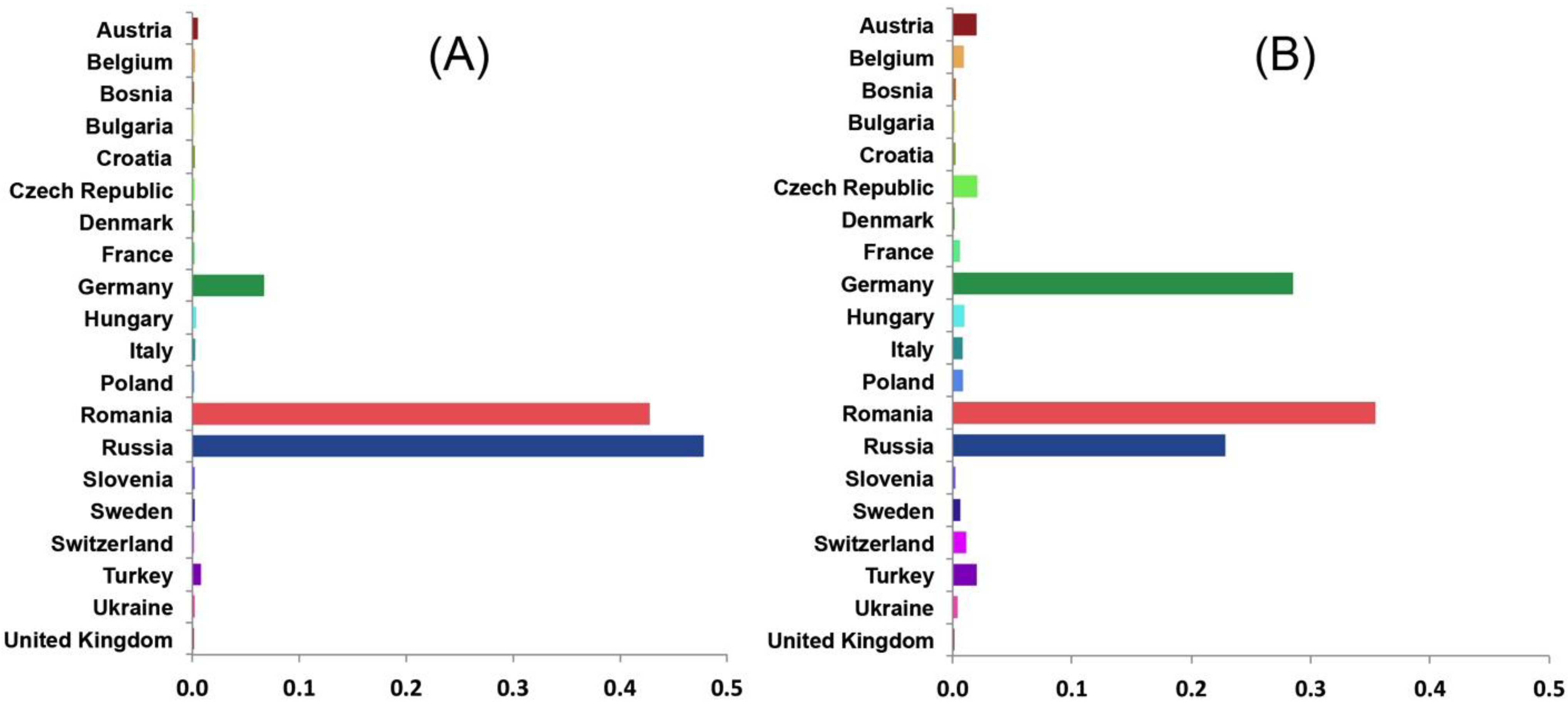
3. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | BF | From | To |
|---|---|---|---|
| HA | 11,049.7 | Wild | Other |
| HA | 11,049.7 | Other | Wild |
| HA | 171.4 | Domestic | Wild |
| NA | 11,049.7 | Wild | Other |
| NA | 11,049.7 | Other | Wild |
| 'NA | 183.0 | Domestic | Wild |




| Tree | Kullback-Leibler | Association Index |
|---|---|---|
| Host Type | ||
| HA | 0.71 | 6.01 (4.97, 7.06) * |
| NA | 0.72 | 7.22 (5.97, 8.44) * |
| Location (by Countries) | ||
| HA | 3.24 | 9.1 (7.87, 10.3) * |
| NA | 1.68 | 10.45 (9.29, 11.58) * |
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kilpatrick, A.M.; Chmura, A.A.; Gibbons, D.W.; Fleischer, R.C.; Marra, P.P.; Daszak, P. Predicting the global spread of H5N1 avian influenza. Proc. Natl. Acad. Sci .USA 2006, 103, 19368–19373. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.H. Summary of avian influenza activity in Europe, Asia, and Africa, 2006–2009. Avian Dis. 2010, 54, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Sims, L.D.; Domenech, J.; Benigno, C.; Kahn, S.; Kamata, A.; Lubroth, J.; Martin, V.; Roeder, P. Origin and evolution of highly pathogenic H5N1 avian influenza in Asia. Vet. Rec. 2005, 157, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Bonn, D. Wild birds, poultry, and avian influenza. Lancet Infect. Dis. 2006, 6, 262–262. [Google Scholar] [CrossRef]
- Xu, X.; Subbarao; Cox, N.J.; Guo, Y. Genetic characterization of the pathogenic influenza A/Goose/Guangdong/1/96 (H5N1) virus: Similarity of its hemagglutinin gene to those of H5N1 viruses from the 1997 outbreaks in Hong Kong. Virology 1999, 261, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, M.M.; Rasool, S.T.; Song, D.; Zhu, C.; Hao, Q.; Zhu, Y.; Wu, J. Origin of highly pathogenic H5N1 avian influenza virus in China and genetic characterization of donor and recipient viruses. J. Gen. Virol. 2007, 88, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- WHO/OIE/FAO H5N1 Evolution Working Group. Toward a unified nomenclature system for highly pathogenic avian influenza virus (H5N1). Emerg. Infect. Dis. 2008, 14. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Li, Z.; Shi, J.; Shinya, K.; Deng, G.; Qi, Q.; Tian, G.; Fan, S.; Zhao, H.; et al. Properties and dissemination of H5N1 viruses isolated during an influenza outbreak in migratory waterfowl in western China. J. Virol. 2006, 80, 5976–5983. [Google Scholar] [CrossRef] [PubMed]
- European Commission (EC). A Report on Surveys for Avian Influenza in Poultry in Member States during 2005. Available online: http://ec.europa.eu/food/animal/diseases/controlmeasures/avian/eu_resp_surveillance_en.htm (accessed on 1 January 2014).
- Globig, A.; Staubach, C.; Beer, M.; Koppen, U.; Fiedler, W.; Nieburg, M.; Wilking, H.; Starick, E.; Teifke, J.P.; Werner, O.; et al. Epidemiological and ornithological aspects of outbreaks of highly pathogenic avian influenza virus H5N1 of Asian lineage in wild birds in Germany, 2006 and 2007. Transbound. Emerg. Dis. 2009, 56, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Harder, T.; Starick, E.; Beer, M.; Werner, O.; Hoffmann, B.; Mettenleiter, T.C.; Mundt, E. Molecular analysis of highly pathogenic avian influenza virus of subtype H5N1 isolated from wild birds and mammals in northern Germany. J. Gen. Virol. 2007, 88, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Starick, E.; Beer, M.; Hoffmann, B.; Staubach, C.; Werner, O.; Globig, A.; Strebelow, G.; Grund, C.; Durban, M.; Conraths, F.J.; et al. Phylogenetic analyses of highly pathogenic avian influenza virus isolates from Germany in 2006 and 2007 suggest at least three separate introductions of H5N1 virus. Vet. Microbiol. 2008, 128, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Breed, A.C.; Harris, K.; Hesterberg, U.; Gould, G.; Londt, B.Z.; Brown, I.H.; Cook, A.J. Surveillance for avian influenza in wild birds in the European Union in 2007. Avian Dis. 2010, 54, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Oprisan, G.; Coste, H.; Lupulescu, E.; Oprisoreanu, A.M.; Szmal, C.; Onita, I.; Popovici, N.; Ionescu, L.E.; Bicheru, S.; Enache, N.; et al. Molecular analysis of the first avian influenza H5N1 isolates from fowl in Romania. Roum. Arch. Microbiol. Immunol. 2006, 65, 79–82. [Google Scholar] [PubMed]
- Hofmann, M.A.; Renzullo, S.; Baumer, A. Phylogenetic characterization of H5N1 highly pathogenic avian influenza viruses isolated in Switzerland in 2006. Virus Genes 2008, 37, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Kiss, I.; Gyarmati, P.; Zohari, S.; Ramsay, K.W.; Metreveli, G.; Weiss, E.; Brytting, M.; Stivers, M.; Lindstrom, S.; Lundkvist, A.; et al. Molecular characterization of highly pathogenic H5N1 avian influenza viruses isolated in Sweden in 2006. Virol. J. 2008, 5. [Google Scholar] [CrossRef] [PubMed]
- Zohari, S.; Gyarmati, P.; Thoren, P.; Czifra, G.; Brojer, C.; Belak, S.; Berg, M. Genetic characterization of the NS gene indicates co-circulation of two sub-lineages of highly pathogenic avian influenza virus of H5N1 subtype in Northern Europe in 2006. Virus Genes 2008, 36, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Vostinakova, V.; Pindova, Z.; Hornickova, J.; Cernikova, L.; Sedlak, K.; Mojzis, M.; Dirbakova, Z.; Machova, J. Molecular and phylogenetic analysis of the H5N1 avian influenza virus caused the first highly pathogenic avian influenza outbreak in poultry in the Czech Republic in 2007. Vet. Microbiol. 2009, 133, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Szeleczky, Z.; Dan, A.; Ursu, K.; Ivanics, E.; Kiss, I.; Erdelyi, K.; Belak, S.; Muller, C.P.; Brown, I.H.; Balint, A. Four different sublineages of highly pathogenic avian influenza H5N1 introduced in Hungary in 2006–2007. Vet. Microbiol. 2009, 139, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Machova, J.; Hornickova, J.; Tomci, M.; Nagl, I.; Horyna, B.; Holko, I. Highly pathogenic avian influenza virus subtype H5N1 in Mute swans in the Czech Republic. Vet. Microbiol. 2007, 120, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F. Bayesian inference of character evolution. Trends Ecol. Evol. 2004, 19, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Jewell, C.P.; Kypraios, T.; Christley, R.M.; Roberts, G.O. A novel approach to real-time risk prediction for emerging infectious diseases: A case study in Avian Influenza H5N1. Prev. Vet. Med. 2009, 91, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Munoz, M.J.; de La Torre, A.; Iglesias, I.; Peris, S.; Infante, O.; Sanchez-Vizcaino, J.M. Risk of introduction of H5N1 HPAI from Europe to Spain by wild water birds in autumn. Transbound. Emerg. Dis. 2009, 56, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.; Fernandez, S.R.; Schobesberger, H.; Koefer, J. Geographical spread of highly pathogenic avian influenza virus H5N1 during the 2006 outbreak in Austria. J. Virol. 2010, 84, 5815–5823. [Google Scholar] [CrossRef] [PubMed]
- Reperant, L.A.; Fuckar, N.S.; Osterhaus, A.D.; Dobson, A.P.; Kuiken, T. Spatial and temporal association of outbreaks of H5N1 influenza virus infection in wild birds with the 0 degrees C isotherm. PLoS Pathog. 2010, 6, e1000854. [Google Scholar] [CrossRef] [PubMed]
- Alkhamis, M.; Willeberg, P.; Carlsson, U.; Carpenter, T.; Perez, A. Alternative scan-based approaches to identify space-time clusters of highly pathogenic avian influenza virus H5N1 in wild birds in Denmark and Sweden in 2006. Avian Dis. 2012, 56, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Minin, V.N.; Suchard, M.A. Fast, accurate and simulation-free stochastic mapping. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3985–3995. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.G.; Fitch, W.M. Influenza A H5N1 immigration is filtered out at some international borders. PLoS ONE 2008, 3, e1697. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.G.; Hodac, H.; Lathrop, R.H.; Fitch, W.M. A statistical phylogeography of influenza A H5N1. Proc. Natl. Acad. Sci. USA 2007, 104, 4473–4478. [Google Scholar] [CrossRef] [PubMed]
- Global Initiative on Sharing All Influenza Data. Available online: http://platform.gisaid.org/epi3/frontend#4ff951 (accessed on 1 December 2013).
- Alkhamis, M.; Perez, A.; Batey, N.; Howard, W.; Watson, S.; Baillie, G.; Franz, S.; Focosi-Snyman, R.; Onita, I.; Cioranu, R.; et al. Modeling the association of space, time, and host species with variation of the HA, NA, and NS genes of H5N1 highly pathogenic avian influenza viruses isolated from birds in Romania in 2005–2007. Avian Dis. 2013, 57, 612–621. [Google Scholar] [CrossRef] [PubMed]
- NCBI Influenza Virus Resource. Available online: http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html (accessed on 15 December 2013).
- Global Animal Disease Information System. Available online: http://empres-i.fao.org/ (accessed on 1 June 2015).
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.P.; Maddison, D.R. Mesquite: A Modular System for Evolutionary Analysis. Version 2.75. 2014. Available online: http://mesquiteproject.org (accessed on 15 March 2014).
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. Partitionfinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Weiss, R.E.; Sinsheimer, J.S. Bayesian selection of continuous-time Markov chain evolutionary models. Mol. Biol. Evol. 2001, 18, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Minin, V.N.; Bloomquist, E.W.; Suchard, M.A. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol. 2008, 25, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Raftery, A.; Newton, M.; Satagopan, J.; Krivitsky, P. Estimating the Integrated Likelihood via Posterior Simulation Using the Harmonic Mean Identity. In Bayesian Statistics 8; Oxford University Press: Oxford, UK, 2007; pp. 371–416. [Google Scholar]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.; Rambaut, A.; Drummond, A.J. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 2006, 23, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Rambaut, A.; Suchard, M.A.; Lemey, P. SPREAD: Spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 2011, 27, 2910–2912. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Lycett, S.J.; Leigh Brown, A.J. Reassortment patterns of avian influenza virus internal segments among different subtypes. BMC Evol. Biol. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Kullback, S.; Leibler, R. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Razavi, N. Kullback-Leibler Divergence. Available online: http://www.mathworks.com/matlabcentral/fileexchange/20688-kullback-leibler-divergence (accessed on 12 September 2014).
- The MathWorks, Inc. Matlab and Statistics Release 2013a. Natick, Massachusettsm United States, 2012. [Google Scholar]
- Parker, J.; Rambaut, A.; Pybus, O.G. Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty. Infect. Genet. Evol. 2008, 8, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.M.; Chernyakhovsky, A.; Rao, V. Modeling and analysis of global epidemiology of avian influenza. Environ. Model. Softw. 2009, 24, 124–134. [Google Scholar] [CrossRef]
- Ward, M.P.; Maftei, D.; Apostu, C.; Suru, A. Geostatistical visualisation and spatial statistics for evaluation of the dispersion of epidemic highly pathogenic avian influenza subtype H5N1. Vet. Res. 2008, 39. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenstrom, J.; Osterhaus, A.D.; Fouchier, R.A. Global patterns of influenza a virus in wild birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Vijaykrishna, D.; Bahl, J.; Riley, S.; Duan, L.; Zhang, J.X.; Chen, H.; Peiris, J.S.; Smith, G.J.; Guan, Y. Evolutionary dynamics and emergence of panzootic H5N1 influenza viruses. PLoS Pathog. 2008, 4, e1000161. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, M.H.; Juan, H.; Jiang, P.; Li, Y.; Li, T.; Du, Y.; Mukhtar, M.M. Complete genome analysis of a highly pathogenic H5N1 influenza A virus isolated from a tiger in China. Arch. Virol. 2008, 153, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Scotch, M.; Sarkar, I.N.; Mei, C.; Leaman, R.; Cheung, K.H.; Ortiz, P.; Singraur, A.; Gonzalez, G. Enhancing phylogeography by improving geographical information from GenBank. J. Biomed. Inform. 2011, 44, S44–S47. [Google Scholar] [CrossRef] [PubMed]
- Tahsin, T.; Beard, R.; Rivera, R.; Lauder, R.; Wallstrom, G.; Scotch, M.; Gonzalez, G. Natural language processing methods for enhancing geographic metadata for phylogeography of zoonotic viruses. AMIA Jt. Summits Transl. Sci. Proc. 2014, 2014, 102–111. [Google Scholar] [PubMed]
- Pybus, O.G.; Fraser, C.; Rambaut, A. Evolutionary epidemiology: Preparing for an age of genomic plenty. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhamis, M.A.; Moore, B.R.; Perez, A.M. Phylodynamics of H5N1 Highly Pathogenic Avian Influenza in Europe, 2005–2010: Potential for Molecular Surveillance of New Outbreaks. Viruses 2015, 7, 3310-3328. https://doi.org/10.3390/v7062773
Alkhamis MA, Moore BR, Perez AM. Phylodynamics of H5N1 Highly Pathogenic Avian Influenza in Europe, 2005–2010: Potential for Molecular Surveillance of New Outbreaks. Viruses. 2015; 7(6):3310-3328. https://doi.org/10.3390/v7062773
Chicago/Turabian StyleAlkhamis, Mohammad A., Brian R. Moore, and Andres M. Perez. 2015. "Phylodynamics of H5N1 Highly Pathogenic Avian Influenza in Europe, 2005–2010: Potential for Molecular Surveillance of New Outbreaks" Viruses 7, no. 6: 3310-3328. https://doi.org/10.3390/v7062773
APA StyleAlkhamis, M. A., Moore, B. R., & Perez, A. M. (2015). Phylodynamics of H5N1 Highly Pathogenic Avian Influenza in Europe, 2005–2010: Potential for Molecular Surveillance of New Outbreaks. Viruses, 7(6), 3310-3328. https://doi.org/10.3390/v7062773