
The European Classical Swine Fever Virus Database: Blueprint for a Pathogen-Specific Sequence Database with Integrated Sequence Analysis Tools


Abstract
:1. Introduction
2. Materials and Methods
2.1. Database (DB) Design
2.2. Nucleotide Sequencing
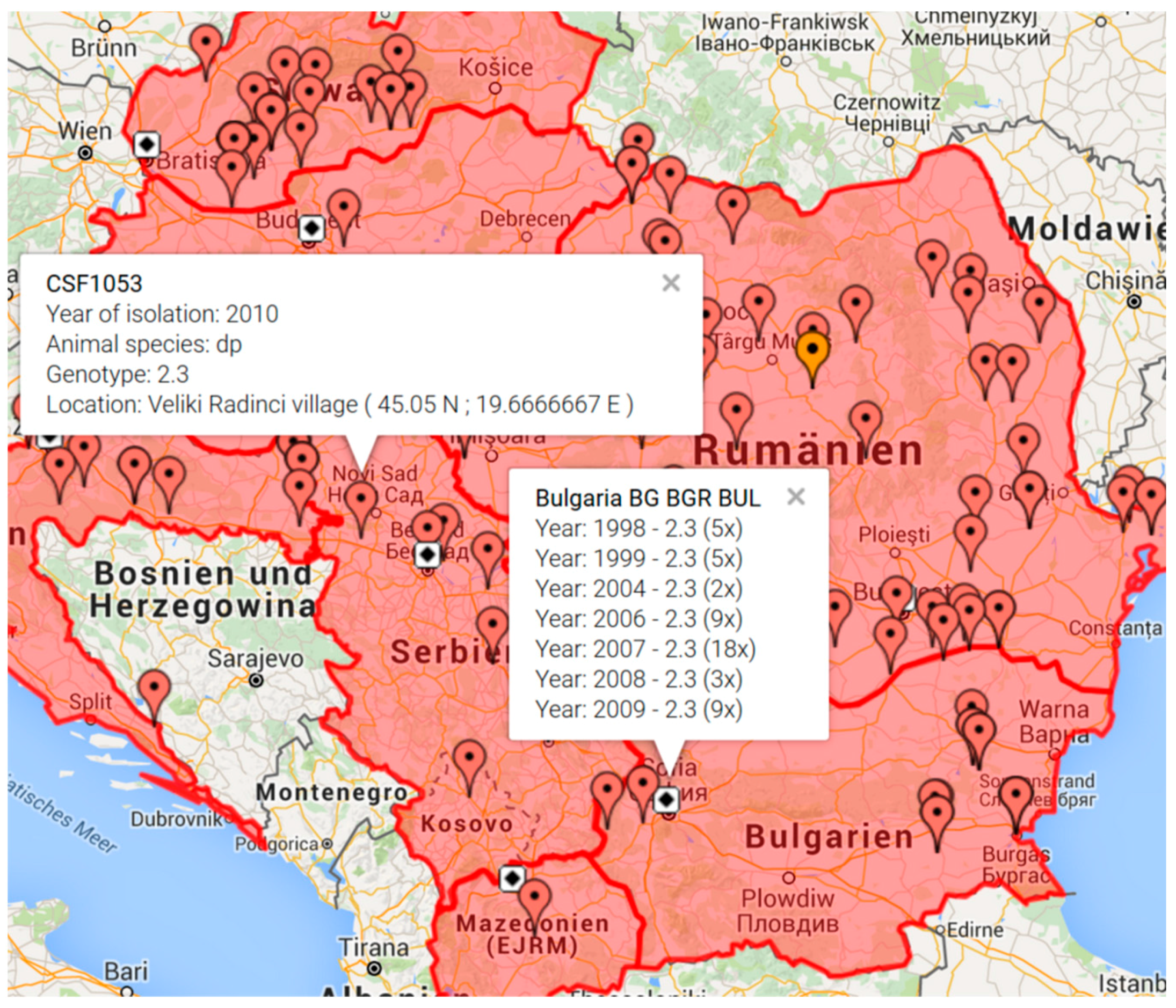
2.3. Implementation of the “CSF Maps” Tool
2.4. Improvements of the Genetic Typing Module
3. Results
3.1. Novel Features and Data Provided by the Classical Swine Fever (CSF) DB (CSF-DB)
3.2. Visualization of outbreak locations by the “CSF Maps” tool
3.3. Novel Features in the Genetic Typing Module
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fischer, N.; Rohde, H.; Indenbirken, D.; Gunther, T.; Reumann, K.; Lutgehetmann, M.; Meyer, T.; Kluge, S.; Aepfelbacher, M.; Alawi, M.; et al. Rapid metagenomic diagnostics for suspected outbreak of severe pneumonia. Emerg. Infect. Dis. 2014, 20, 1072–1075. [Google Scholar] [CrossRef]
- Franca, L.T.; Carrilho, E.; Kist, T.B. A review of DNA sequencing techniques. Q. Rev. Biophys. 2002, 35, 169–200. [Google Scholar] [CrossRef]
- Leifer, I.; Hoffmann, B.; Hoper, D.; Bruun Rasmussen, T.; Blome, S.; Strebelow, G.; Horeth-Bontgen, D.; Staubach, C.; Beer, M. Molecular epidemiology of current Classical Swine Fever virus isolates of wild boar in germany. J. Gen. Virol. 2010, 91, 2687–2697. [Google Scholar] [CrossRef]
- Barzon, L.; Lavezzo, E.; Costanzi, G.; Franchin, E.; Toppo, S.; Palu, G. Next-generation sequencing technologies in diagnostic virology. J. Clin. Virol. 2013, 58, 346–350. [Google Scholar] [CrossRef]
- Faure, D.; Joly, D. Next-generation sequencing as a powerful motor for advances in the biological and environmental sciences. Genetica 2015, 143, 129–132. [Google Scholar] [CrossRef]
- World Organisation for Animal Health (OIE). Resolutions. In Proceedings of the 83rd General Session of the World Assembly of OIE Delegates, Paris, France, 24–29 May 2015; pp. 129–194.
- Dreier, S.; Zimmermann, B.; Moennig, V.; Greiser-Wilke, I. A sequence database allowing automated genotyping of Classical Swine Fever virus isolates. J. Virol. Methods 2007, 140, 95–99. [Google Scholar] [CrossRef]
- Greiser-Wilke, I.; Zimmermann, B.; Fritzemeier, J.; Floegel, G.; Moennig, V. Structure and presentation of a world wide web database of csf virus isolates held at the eu reference laboratory. Vet. Microbiol. 2000, 73, 131–136. [Google Scholar] [CrossRef]
- Paton, D.J.; McGoldrick, A.; Greiser-Wilke, I.; Parchariyanon, S.; Song, J.Y.; Liou, P.P.; Stadejek, T.; Lowings, J.P.; Bjorklund, H.; Belak, S. Genetic typing of Classical Swine Fever virus. Vet. Microbiol. 2000, 73, 137–157. [Google Scholar] [CrossRef]
- Postel, A.; Jha, V.C.; Schmeiser, S.; Becher, P. First molecular identification and characterization of Classical Swine Fever virus isolates from nepal. Arch. Virol. 2013, 158, 207–210. [Google Scholar] [CrossRef]
- Postel, A.; Schmeiser, S.; Bernau, J.; Meindl-Boehmer, A.; Pridotkas, G.; Dirbakova, Z.; Mojzis, M.; Becher, P. Improved strategy for phylogenetic analysis of Classical Swine Fever virus based on full-length e2 encoding sequences. Vet. Res. 2012, 43, 50. [Google Scholar] [CrossRef]
- Postel, A.; Schmeiser, S.; Perera, C.L.; Rodriguez, L.J.; Frias-Lepoureau, M.T.; Becher, P. Classical Swine Fever virus isolates from cuba form a new subgenotype 1.4. Vet. Microbiol. 2013, 161, 334–338. [Google Scholar] [CrossRef]
- Anonymous. Developer’s Guide, Google Maps Geocoding API, Google Developers. Available online: https://developers.Google.Com/maps/documentation/geocoding/intro (accessed on 23 April 2012).
- Anonymous. Google Maps JavaScript API, Google Developers. Available online: https://developers.Google.Com/maps/documentation/javascript/ (accessed on 23 April 2012).
- Anonymous. Northrop Grumman Health IT, J. Craig Venter Institute, and Vecna Technologies. Available online: https://www.Fludb.Org (accessed on 19 August 2016).
- Anonymous. Epiflu db, Freunde von GISAID e.V. Available online: https://www.Platform.Gisaid.Org (accessed on 19 August 2016).
- Stalder, H.; Hug, C.; Zanoni, R.; Vogt, H.R.; Peterhans, E.; Schweizer, M.; Bachofen, C. A nationwide database linking information on the hosts with sequence data of their virus strains: A useful tool for the eradication of bovine viral diarrhea (bvd) in switzerland. Virus Res. 2016, 218, 49–56. [Google Scholar] [CrossRef]
- Pickett, B.E.; Greer, D.S.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.; Gu, Z.; Kumar, S.; Zaremba, S.; Larsen, C.N.; et al. Virus pathogen database and analysis resource (vipr): A comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses 2012, 4, 3209–3226. [Google Scholar] [CrossRef]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. Vipr: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Lam, T.T.; Hon, C.C.; Tang, J.W. Use of phylogenetics in the molecular epidemiology and evolutionary studies of viral infections. Crit. Rev. Clin. Lab. Sci. 2010, 47, 5–49. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Genome Region | 5′NTR | E2 Fragment | E2 Gene | NS5B | 5′NTR-E2 | Full Genome |
|---|---|---|---|---|---|---|
| (Nucleotides) | (150) | (190) | (1119) | (409) * | (3.3 k) * | (12.3 k) * |
| Isolates (CSF No.) | 661 | 557 | 171 | 43 | 46 | 11 |
| only sequence (XXX No.) | 251 | 525 | 94 | 96 | 44 | 42 |
| Total | 912 | 1082 | 265 | 139 | 90 | 53 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Postel, A.; Schmeiser, S.; Zimmermann, B.; Becher, P. The European Classical Swine Fever Virus Database: Blueprint for a Pathogen-Specific Sequence Database with Integrated Sequence Analysis Tools. Viruses 2016, 8, 302. https://doi.org/10.3390/v8110302
Postel A, Schmeiser S, Zimmermann B, Becher P. The European Classical Swine Fever Virus Database: Blueprint for a Pathogen-Specific Sequence Database with Integrated Sequence Analysis Tools. Viruses. 2016; 8(11):302. https://doi.org/10.3390/v8110302
Chicago/Turabian StylePostel, Alexander, Stefanie Schmeiser, Bernd Zimmermann, and Paul Becher. 2016. "The European Classical Swine Fever Virus Database: Blueprint for a Pathogen-Specific Sequence Database with Integrated Sequence Analysis Tools" Viruses 8, no. 11: 302. https://doi.org/10.3390/v8110302
APA StylePostel, A., Schmeiser, S., Zimmermann, B., & Becher, P. (2016). The European Classical Swine Fever Virus Database: Blueprint for a Pathogen-Specific Sequence Database with Integrated Sequence Analysis Tools. Viruses, 8(11), 302. https://doi.org/10.3390/v8110302