
Next-Generation Sequencing in the Understanding of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Biology



Abstract
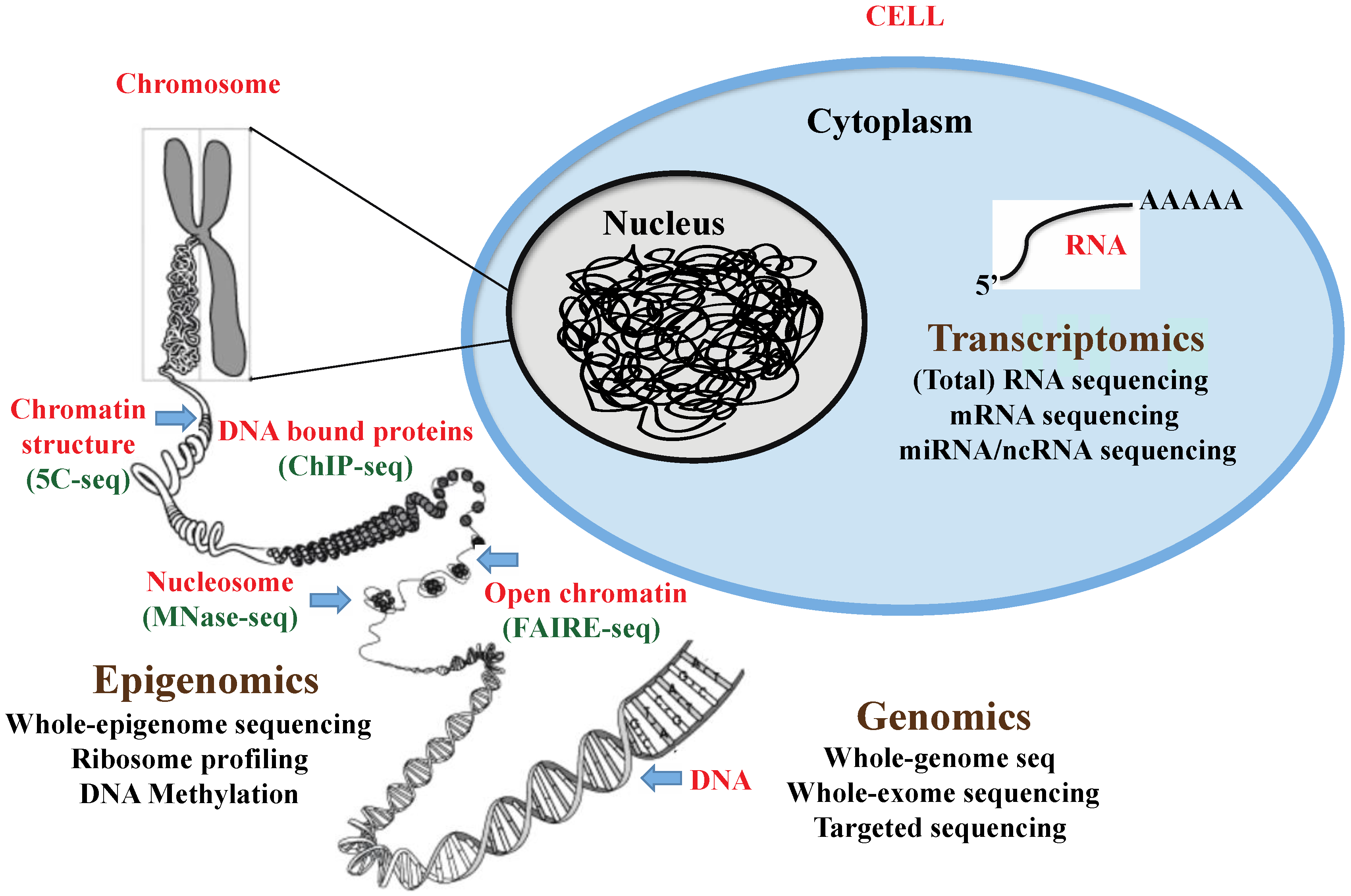
:1. Introduction
2. Unlocking the KSHV Genome and Insights into KSHV Infection from Host Genomics: The Contributions of Whole-Genome Sequencing, Whole-Exome-Sequencing and Targeted-Sequencing
3. An Atlas of the KSHV Transcriptome: The Contributions of RNA-Sequencing and Small RNA/Non-Coding RNA-Sequencing
3.1. RNA-Sequencing
3.2. Genome-Wide Analysis of the Latent and Lytic KSHV Transcriptomes
3.3. Small RNA/Non-Coding RNA-Sequencing
3.3.1. Role of miRNAs in KSHV Latency and Lytic Reactivation
3.3.2. Role of ncRNAs in KSHV Lytic Reactivation
4. Whole-KSHV Epigenome-Mapping: The Contributions of DNA Methylation-Sequencing, ChIP-Sequencing and FAIRE-Sequencing
4.1. Sequencing of the KSHV Epigenome during de Novo Infection and Latency
4.2. Sequencing of the KSHV and Host Epigenomes during Lytic Reactivation and Replication
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dourmishev, L.A.; Dourmishev, A.L.; Palmeri, D.; Schwartz, R.A.; Lukac, D.M. Molecular genetics of kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 2003, 67, 175–212. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Robertson, E.S. Molecular biology and pathogenesis of kaposi sarcoma-associated herpesvirus. FEMS Microbiol. Lett. 2003, 222, 155–163. [Google Scholar] [CrossRef]
- Sarda, S.; Hannenhalli, S. Next-generation sequencing and epigenomics research: A hammer in search of nails. Genom. Inform. 2014, 12, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in aids-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in aids-associated kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Boshoff, C.; Weiss, R. Aids-related malignancies. Nat. Rev. Cancer 2002, 2, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Dukers, N.H.; Rezza, G. Human herpesvirus 8 epidemiology: What we do and do not know. Aids 2003, 17, 1717–1730. [Google Scholar] [CrossRef] [PubMed]
- Kedes, D.H.; Operskalski, E.; Busch, M.; Kohn, R.; Flood, J.; Ganem, D. The seroepidemiology of human herpesvirus 8 (kaposi’s sarcoma-associated herpesvirus): Distribution of infection in KS risk groups and evidence for sexual transmission. Nat. Med. 1996, 2, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.C.; Ciufo, D.M.; Alcendor, D.J.; Wan, X.; Nicholas, J.; Browning, P.J.; Rady, P.L.; Tyring, S.K.; Orenstein, J.M.; Rabkin, C.S.; et al. High-level variability in the ORF-K1 membrane protein gene at the left end of the kaposi’s sarcoma-associated herpesvirus genome defines four major virus subtypes and multiple variants or clades in different human populations. J. Virol. 1999, 73, 4156–4170. [Google Scholar] [PubMed]
- Poole, L.J.; Zong, J.C.; Ciufo, D.M.; Alcendor, D.J.; Cannon, J.S.; Ambinder, R.; Orenstein, J.M.; Reitz, M.S.; Hayward, G.S. Comparison of genetic variability at multiple loci across the genomes of the major subtypes of kaposi’s sarcoma-associated herpesvirus reveals evidence for recombination and for two distinct types of open reading frame K15 alleles at the right-hand end. J. Virol. 1999, 73, 6646–6660. [Google Scholar] [PubMed]
- Hayward, G.S.; Zong, J.C. Modern evolutionary history of the human KSHV genome. Curr. Top. Microbiol. Immunol. 2007, 312, 1–42. [Google Scholar] [PubMed]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.; Marra, M.A. Applications of next-generation sequencing technologies in functional genomics. Genomics 2008, 92, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Neipel, F.; Albrecht, J.C.; Fleckenstein, B. Cell-homologous genes in the kaposi’s sarcoma-associated rhadinovirus human herpesvirus 8: Determinants of its pathogenicity? J. Virol. 1997, 71, 4187–4192. [Google Scholar] [PubMed]
- Rezaee, S.A.; Cunningham, C.; Davison, A.J.; Blackbourn, D.J. Kaposi’s sarcoma-associated herpesvirus immune modulation: An overview. J. Gen. Virol. 2006, 87, 1781–1804. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.C.; Zhang, Y.J.; Deng, J.H.; Wang, X.P.; Pan, H.Y.; Hettler, E.; Gao, S.J. Efficient infection by a recombinant kaposi’s sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: Application for genetic analysis. J. Virol. 2002, 76, 6185–6196. [Google Scholar] [CrossRef] [PubMed]
- Yakushko, Y.; Hackmann, C.; Gunther, T.; Ruckert, J.; Henke, M.; Koste, L.; Alkharsah, K.; Bohne, J.; Grundhoff, A.; Schulz, T.F.; et al. Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome contains a duplication of a long unique-region fragment within the terminal repeat region. J. Virol. 2011, 85, 4612–4617. [Google Scholar] [CrossRef] [PubMed]
- Brulois, K.F.; Chang, H.; Lee, A.S.; Ensser, A.; Wong, L.Y.; Toth, Z.; Lee, S.H.; Lee, H.R.; Myoung, J.; Ganem, D.; et al. Construction and manipulation of a new kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 2012, 86, 9708–9720. [Google Scholar] [CrossRef] [PubMed]
- Tamburro, K.M.; Yang, D.; Poisson, J.; Fedoriw, Y.; Roy, D.; Lucas, A.; Sin, S.-H.; Malouf, N.; Moylan, V.; Damania, B.; et al. Vironome of kaposi sarcoma associated herpesvirus-inflammatory cytokine syndrome in an aids patient reveals co-infection of human herpesvirus 8 and human herpesvirus 6a. Virology 2012, 433, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Olp, L.N.; Jeanniard, A.; Marimo, C.; West, J.T.; Wood, C. Whole-genome sequencing of kaposi’s sarcoma-associated herpesvirus from zambian kaposi’s sarcoma biopsy specimens reveals unique viral diversity. J. Virol. 2015, 89, 12299–12308. [Google Scholar] [PubMed]
- Bruce, A.G.; Ryan, J.T.; Thomas, M.J.; Peng, X.; Grundhoff, A.; Tsai, C.C.; Rose, T.M. Next-generation sequence analysis of the genome of RFHVMn, the macaque homolog of kaposi’s sarcoma (KS)-associated herpesvirus, from a KS-like tumor of a pig-tailed macaque. J. Virol. 2013, 87, 13676–13693. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Palser, A.L.; Watson, S.J.; Lai, I.Y.; Gray, E.R.; Grant, P.; Kanda, R.K.; Leproust, E.; Kellam, P.; Breuer, J. Specific capture and whole-genome sequencing of viruses from clinical samples. PLoS ONE 2011, 6, e27805. [Google Scholar] [CrossRef] [PubMed]
- Byun, M.; Abhyankar, A.; Lelarge, V.; Plancoulaine, S.; Palanduz, A.; Telhan, L.; Boisson, B.; Picard, C.; Dewell, S.; Zhao, C.; et al. Whole-exome sequencing-based discovery of stim1 deficiency in a child with fatal classic kaposi sarcoma. J. Exp. Med. 2010, 207, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Byun, M.; Ma, C.S.; Akcay, A.; Pedergnana, V.; Palendira, U.; Myoung, J.; Avery, D.T.; Liu, Y.; Abhyankar, A.; Lorenzo, L.; et al. Inherited human OX40 deficiency underlying classic kaposi sarcoma of childhood. J. Exp. Med. 2013, 210, 1743–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Aavikko, M.; Kaasinen, E.; Nieminen, J.K.; Byun, M.; Donner, I.; Mancuso, R.; Ferrante, P.; Clerici, M.; Brambilla, L.; Tourlaki, A.; et al. Whole-genome sequencing identifies STAT4 as a putative susceptibility gene in classic kaposi sarcoma. J. Infect. Dis. 2015, 211, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, W.; Xiong, J.; Sherrod, C.J.; Henry, D.H.; Dittmer, D.P. Interleukin 1 receptor-associated kinase 1 (IRAK1) mutation is a common, essential driver for kaposi sarcoma herpesvirus lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E4762–E4768. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.S.; Ciufo, D.; Hawkins, A.L.; Griffin, C.A.; Borowitz, M.J.; Hayward, G.S.; Ambinder, R.F. A new primary effusion lymphoma-derived cell line yields a highly infectious kaposi’s sarcoma herpesvirus-containing supernatant. J. Virol. 2000, 74, 10187–10193. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Ganem, D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in kaposi sarcoma pathogenesis. J. Clin. Investig. 2004, 113, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Zhong, W.; Herndier, B.; McGrath, M.; Abbey, N.; Kedes, D.; Ganem, D. Lytic growth of kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 1996, 2, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Sugden, B. New viruses shake old paradigms. J. Clin. Investig. 2004, 113, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Lagunoff, M.; Renne, R.; Staskus, K.; Haase, A.; Ganem, D. A cluster of latently expressed genes in kaposi’s sarcoma-associated herpesvirus. J. Virol. 1998, 72, 8309–8315. [Google Scholar] [PubMed]
- Fakhari, F.D.; Dittmer, D.P. Charting latency transcripts in kaposi’s sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 2002, 76, 6213–6223. [Google Scholar] [CrossRef] [PubMed]
- Samols, M.A.; Hu, J.; Skalsky, R.L.; Renne, R. Cloning and identification of a microrna cluster within the latency-associated region of kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 9301–9305. [Google Scholar] [CrossRef] [PubMed]
- Sarid, R.; Flore, O.; Bohenzky, R.A.; Chang, Y.; Moore, P.S. Transcription mapping of the kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 1998, 72, 1005–1012. [Google Scholar] [PubMed]
- Jeong, J.; Papin, J.; Dittmer, D. Differential regulation of the overlapping kaposi’s sarcoma-associated herpesvirus vgcr (ORF74) and lana (ORF73) promoters. J. Virol. 2001, 75, 1798–1807. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zeng, Y.; Huang, Z.; Huang, L.; Qian, C.; Tang, G.; Qin, D. Human herpesvirus 6 activates lytic cycle replication of kaposi’s sarcoma-associated herpesvirus. Am. J. Pathol. 2005, 166, 173–183. [Google Scholar] [CrossRef]
- Mercader, M.; Taddeo, B.; Panella, J.R.; Chandran, B.; Nickoloff, B.J.; Foreman, K.E. Induction of HHV-8 lytic cycle replication by inflammatory cytokines produced by HIV-1-infected T cells. Am. J. Pathol. 2000, 156, 1961–1971. [Google Scholar] [CrossRef]
- Vieira, J.; O’Hearn, P.; Kimball, L.; Chandran, B.; Corey, L. Activation of kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) lytic replication by human cytomegalovirus. J. Virol. 2001, 75, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhou, F.; Bedolla, R.G.; Jones, T.; Lei, X.; Kang, T.; Guadalupe, M.; Gao, S.J. Reactive oxygen species hydrogen peroxide mediates kaposi’s sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 2011, 7, e1002054. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.A.; Rinderknecht, A.S.; Zoeteweij, J.P.; Aoki, Y.; Read-Connole, E.L.; Tosato, G.; Blauvelt, A.; Yarchoan, R. Hypoxia induces lytic replication of kaposi sarcoma-associated herpesvirus. Blood 2001, 97, 3244–3250. [Google Scholar] [CrossRef] [PubMed]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [PubMed]
- Sun, R.; Lin, S.F.; Staskus, K.; Gradoville, L.; Grogan, E.; Haase, A.; Miller, G. Kinetics of kaposi’s sarcoma-associated herpesvirus gene expression. J. Virol. 1999, 73, 2232–2242. [Google Scholar] [PubMed]
- Zhu, F.X.; Cusano, T.; Yuan, Y. Identification of the immediate-early transcripts of kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5556–5567. [Google Scholar] [PubMed]
- Jenner, R.G.; Alba, M.M.; Boshoff, C.; Kellam, P. Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 2001, 75, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Kirshner, J.R.; Ganem, D. Transcriptional activation by the product of open reading frame 50 of kaposi’s sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 1999, 73, 9348–9361. [Google Scholar] [PubMed]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Chandriani, S.; Xu, Y.; Ganem, D. The lytic transcriptome of kaposi’s sarcoma-associated herpesvirus reveals extensive transcription of noncoding regions, including regions antisense to important genes. J. Virol. 2010, 84, 7934–7942. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.P. Transcription profile of kaposi’s sarcoma-associated herpesvirus in primary kaposi’s sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 2003, 63, 2010–2015. [Google Scholar] [PubMed]
- Dresang, L.R.; Teuton, J.R.; Feng, H.; Jacobs, J.M.; Camp, D.G., 2nd; Purvine, S.O.; Gritsenko, M.A.; Li, Z.; Smith, R.D.; Sugden, B.; et al. Coupled transcriptome and proteome analysis of human lymphotropic tumor viruses: Insights on the detection and discovery of viral genes. BMC Genom. 2011, 12. [Google Scholar] [CrossRef]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Liang, D.; Lan, K. Micrornas and unusual small rnas discovered in kaposi’s sarcoma-associated herpesvirus virions. J. Virol. 2012, 86, 12717–12730. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, J.; Grundhoff, A.; Ganem, D. RNAs in the virion of kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 10138–10146. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, P.; Thakker, S.; Verma, S.C. Transcriptome analysis of kaposi’s sarcoma-associated herpesvirus during de novo primary infection of human B and endothelial cells. J. Virol. 2015, 89, 3093–3111. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Day, L.; Gao, S.J.; Lieberman, P.M. Acetylation of the latency-associated nuclear antigen regulates repression of kaposi’s sarcoma-associated herpesvirus lytic transcription. J. Virol. 2006, 80, 5273–5282. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liang, D.; Gao, Y.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J. Virol. 2014, 88, 7331–7344. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Cai, S.; Zhu, C.; Verma, S.C.; Choi, J.Y.; Robertson, E.S. A unique sumo-2-interacting motif within lana is essential for KSHV latency. PLoS Pathog. 2013, 9, e1003750. [Google Scholar] [CrossRef] [PubMed]
- Gjyshi, O.; Roy, A.; Dutta, S.; Veettil, M.V.; Dutta, D.; Chandran, B. Activated NRF2 interacts with kaposi’s sarcoma-associated herpesvirus latency protein LANA-1 and host protein KAP1 to mediate global lytic gene repression. J. Virol. 2015, 89, 7874–7892. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Li, D.J.; Krueger, B.; Renne, R.; Swaminathan, S. Identification of the physiological gene targets of the essential lytic replicative kaposi’s sarcoma-associated herpesvirus ORF57 protein. J. Virol. 2015, 89, 1688–1702. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Wu, N.C.; Xie, Y.; Feng, J.; Tong, L.; Brulois, K.F.; Luan, H.; Du, Y.; Jung, J.U.; Wang, C.-Y.; et al. Kaposi’s sarcoma-associated herpesvirus ORF18 and ORF30 are essential for late gene expression during lytic replication. J. Virol. 2014, 88, 11369–11382. [Google Scholar] [CrossRef] [PubMed]
- Davis, Z.H.; Hesser, C.R.; Park, J.; Glaunsinger, B.A. Interaction between ORF24 and ORF34 in the kaposi’s sarcoma-associated herpesvirus late gene transcription factor complex is essential for viral late gene expression. J. Virol. 2015, 90, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Aubry, V.; Mure, F.; Mariame, B.; Deschamps, T.; Wyrwicz, L.S.; Manet, E.; Gruffat, H. Epstein-barr virus late gene transcription depends on the assembly of a virus-specific preinitiation complex. J. Virol. 2014, 88, 12825–12838. [Google Scholar] [CrossRef] [PubMed]
- Davis, Z.H.; Verschueren, E.; Jang, G.M.; Kleffman, K.; Johnson, J.R.; Park, J.; von Dollen, J.; Maher, M.C.; Johnson, T.; Newton, W.; et al. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol. Cell 2015, 57, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mrna turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [CrossRef]
- Glaunsinger, B.; Chavez, L.; Ganem, D. The exonuclease and host shutoff functions of the SOX protein of kaposi’s sarcoma-associated herpesvirus are genetically separable. J. Virol. 2005, 79, 7396–7401. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, S.; Gaglia, M.M.; Kumar, G.R.; Wong, W.; Jackson, A.O.; Glaunsinger, B.A. Coordinated destruction of cellular messages in translation complexes by the gammaherpesvirus host shutoff factor and the mammalian exonuclease XRN1. PLoS Pathog. 2011, 7, e1002339. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, E.; Gilbertson, S.; Alla, R.; Glaunsinger, B. Viral nucleases induce an mRNA degradation-transcription feedback loop in mammalian cells. Cell Host Microbe 2015, 18, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Nishi, J.; Arimura, K.; Utsunomiya, A.; Yonezawa, S.; Kawakami, K.; Maeno, N.; Ijichi, O.; Ikarimoto, N.; Nakata, M.; Kitajima, I.; et al. Expression of vascular endothelial growth factor in sera and lymph nodes of the plasma cell type of castleman’s disease. Br. J. Haematol. 1999, 104, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Ganem, D. Highly selective escape from KSHV-mediated host mrna shutoff and its implications for viral pathogenesis. J. Exp. Med. 2004, 200, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Clyde, K.; Glaunsinger, B.A. Deep sequencing reveals direct targets of gammaherpesvirus-induced mRNA decay and suggests that multiple mechanisms govern cellular transcript escape. PLoS ONE 2011, 6, e19655. [Google Scholar] [CrossRef] [PubMed]
- Gaglia, M.M.; Rycroft, C.H.; Glaunsinger, B.A. Transcriptome-wide cleavage site mapping on cellular mrnas reveals features underlying sequence-specific cleavage by the viral ribonuclease SOX. PLoS Pathog. 2015, 11, e1005305. [Google Scholar] [CrossRef] [PubMed]
- Sei, E.; Wang, T.; Hunter, O.V.; Xie, Y.; Conrad, N.K. HITS-CLIP analysis uncovers a link between the kaposi’s sarcoma-associated herpesvirus ORF57 protein and host pre-mRNA metabolism. PLoS Pathog. 2015, 11, e1004652. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Strong, M.J.; Wang, X.; Moss, W.N.; Concha, M.; Lin, Z.; O’Grady, T.; Baddoo, M.; Fewell, C.; Renne, R.; et al. High-throughput RNA sequencing-based virome analysis of 50 lymphoma cell lines from the cancer cell line encyclopedia project. J. Virol. 2015, 89, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lei, X.; Bai, Z.; Ye, F.; Huang, Y.; Gao, S.J. Regulation of herpesvirus lifecycle by viral micrornas. Virulence 2010, 1, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Jakymiw, A.; Findlay, V.; Parsons, C. KSHV-encoded microRNAs: Lessons for viral cancer pathogenesis and emerging concepts. Int. J. Cell Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kowdley, K.V. MicroRNAs in common human diseases. Genom. Proteom. Bioinf. 2012, 10, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E. Kaposi’s sarcoma-associated herpesvirus microRNAs. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, D.; Kieffer-Kwon, P.; Ziegelbauer, J.M. Emerging themes from EBV and KSHV microRNA targets. Viruses 2012, 4, 1687–1710. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Cullen, B.R. Transcriptional origin of kaposi’s sarcoma-associated herpesvirus microRNAs. J. Virol. 2006, 80, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods. 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [PubMed]
- Marshall, V.; Parks, T.; Bagni, R.; Wang, C.D.; Samols, M.A.; Hu, J.; Wyvil, K.M.; Aleman, K.; Little, R.F.; Yarchoan, R.; et al. Conservation of virally encoded microRNAs in kaposi sarcoma-associated herpesvirus in primary effusion lymphoma cell lines and in patients with kaposi sarcoma or multicentric castleman disease. J. Infect. Dis. 2007, 195, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Sullivan, C.S.; Ganem, D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 2006, 12, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Cullen, B.R. In-depth analysis of kaposi’s sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. J. Virol. 2010, 84, 695–703. [Google Scholar] [CrossRef] [PubMed]
- AuCoin, D.P.; Colletti, K.S.; Xu, Y.; Cei, S.A.; Pari, G.S. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) contains two functional lytic origins of DNA replication. J. Virol. 2002, 76, 7890–7896. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Li, H.; Wang, Y.; Zhu, F.X.; Kudchodkar, S.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication: Identification of the ori-Lyt and association of K8 bZip protein with the origin. J. Virol. 2003, 77, 5578–5588. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Kincaid, R.P.; Arasappan, D.; Dowd, S.E.; Hunicke-Smith, S.P.; Sullivan, C.S. Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. RNA 2010, 16, 1540–1558. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Peruzzi, F.; Reiss, K.; Dai, L. Role of host microRNAs in kaposi’s sarcoma-associated herpesvirus pathogenesis. Viruses 2014, 6, 4571–4580. [Google Scholar] [CrossRef] [PubMed]
- Boss, I.W.; Plaisance, K.B.; Renne, R. Role of virus-encoded microRNAs in herpesvirus biology. Trends Microbiol. 2009, 17, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Lee, S.; Chaudhary, P.M.; Gill, P.; Jung, J.U. Immune evasion by kaposi’s sarcoma-associated herpesvirus. Future Microbiol. 2010, 5, 1349–1365. [Google Scholar] [CrossRef] [PubMed]
- Moody, R.; Zhu, Y.; Huang, Y.; Cui, X.; Jones, T.; Bedolla, R.; Lei, X.; Bai, Z.; Gao, S.J. KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog. 2013, 9, e1003857. [Google Scholar] [CrossRef] [PubMed]
- Haecker, I.; Renne, R. HITS-CLIP and PAR-CLIP advance viral miRNA targetome analysis. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Haecker, I.; Gay, L.A.; Yang, Y.; Hu, J.; Morse, A.M.; McIntyre, L.M.; Renne, R. Ago HITS-CLIP expands understanding of kaposi’s sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog. 2012, 8, e1002884. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Corcoran, D.L.; Mukherjee, N.; Skalsky, R.L.; Hafner, M.; Nusbaum, J.D.; Shamulailatpam, P.; Love, C.L.; Dave, S.S.; Tuschl, T.; et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 2011, 10, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Viollet, C.; Davis, D.A.; Reczko, M.; Ziegelbauer, J.M.; Pezzella, F.; Ragoussis, J.; Yarchoan, R. Next-generation sequencing analysis reveals differential expression profiles of miRNA-mRNA target pairs in KSHV-infected cells. PLoS ONE 2015, 10, e0126439. [Google Scholar]
- Shaw, G.; Kamen, R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 1986, 46, 659–667. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef] [PubMed]
- McClure, L.V.; Kincaid, R.P.; Burke, J.M.; Grundhoff, A.; Sullivan, C.S. Comprehensive mapping and analysis of kaposi’s sarcoma-associated herpesvirus 3′ UTRs identify differential posttranscriptional control of gene expression in lytic vs. latent infection. J. Virol. 2013, 87, 12838–12849. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Ni, T.; Yang, W.; Meng, B.; Zhu, J.; Zheng, Z.M. A viral genome landscape of RNA polyadenylation from KSHV latent to lytic infection. PLoS Pathog. 2013, 9, e1003749. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Huang, Y.; Li, W.; Zhu, Y.; Jung, J.U.; Lu, C.; Gao, S.J. Genomewide mapping and screening of kaposi’s sarcoma-associated herpesvirus (KSHV) 3′ untranslated regions identify bicistronic and polycistronic viral transcripts as frequent targets of KSHV microRNAs. J. Virol. 2014, 88, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Huang, Y.; Jung, J.U.; Lu, C.; Gao, S.J. Viral miRNA targeting of bicistronic and polycistronic transcripts. Curr. Opin. Virol. 2014, 7, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Wang, V.; Davis, D.A.; Zheng, Z.M.; Yarchoan, R. Genetic organization and hypoxic activation of the kaposi’s sarcoma-associated herpesvirus ORF34-37 gene cluster. J. Virol. 2006, 80, 7037–7051. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Zhou, F.; Lei, X.; Ma, X.; Lu, C.; Gao, S.J. A cluster of transcripts encoded by KSHV ORF30-33 gene locus. Virus Genes 2012, 44, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Bellare, P.; Ganem, D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: An evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 2009, 6, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liang, D.; He, Z.; Deng, Q.; Robertson, E.S.; Lan, K. miR-K12-7-5p encoded by kaposi’s sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS ONE 2011, 6, e16224. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.R.; Ganem, D. Viral microRNA target allows insight into the role of translation in governing microrna target accessibility. Proc. Natl. Acad. Sci. USA 2011, 108, 5148–5153. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G.S. PAN’S labyrinth: Molecular biology of kaposi’s sarcoma-associated herpesvirus (KSHV) PAN RNA, a multifunctional long noncoding RNA. Viruses 2014, 6, 4212–4226. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Kung, H.J.; Izumiya, Y. Long non-coding RNA and epigenetic gene regulation of KSHV. Viruses 2014, 6, 4165–4177. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.-F.; Gradoville, L.; Miller, G. Polyadenylylated nuclear RNA encoded by kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1996, 93, 11883–11888. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Tarrant-Elorza, M.; Verma, S.; Purushothaman, P.; Pari, G.S. Regulation of viral and cellular gene expression by kaposi’s sarcoma-associated herpesvirus polyadenylated nuclear RNA. J. Virol. 2013, 87, 5540–5553. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 2012, 8, e1002680. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Darricarrere, N.; Darnell, A.; Myoung, J.; Steitz, J.A. A viral nuclear noncoding RNA binds re-localized poly(a) binding protein and is required for late KSHV gene expression. PLoS Pathog. 2011, 7, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Kim, K.Y.; Chang, P.C.; Huerta, S.; Shevchenko, B.; Wang, D.H.; Izumiya, C.; Kung, H.J.; Izumiya, Y. A lytic viral long noncoding RNA modulates the function of a latent protein. J. Virol. 2014, 88, 1843–1848. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Brown, H.J.; Wu, T.T.; Sun, R. Transcription activation of polyadenylated nuclear RNA by Rta in human herpesvirus 8/kaposi’s sarcoma-associated herpesvirus. J. Virol. 2001, 75, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Shedd, D.; Gradoville, L.; Cho, M.S.; Chen, L.W.; Chang, J.; Miller, G. Open reading frame 50 protein of kaposi’s sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 2002, 76, 3168–3178. [Google Scholar] [CrossRef] [PubMed]
- Massimelli, M.J.; Kang, J.G.; Majerciak, V.; Le, S.Y.; Liewehr, D.J.; Steinberg, S.M.; Zheng, Z.M. Stability of a long noncoding viral RNA depends on a 9-nt core element at the RNA 5′ end to interact with viral ORF57 and cellular PABPC1. Int. J. Biol. Sci. 2011, 7, 1145–1160. [Google Scholar] [CrossRef] [PubMed]
- Sahin, B.B.; Patel, D.; Conrad, N.K. Kaposi’s sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog. 2010, 6, e1000799. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Jaber, T.; Yuan, Y. A virally encoded small peptide regulates RTA stability and facilitates kaposi’s sarcoma-associated herpesvirus lytic replication. J. Virol. 2013, 87, 3461–3470. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. Chip-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Gunther, T.; Grundhoff, A. The epigenetic landscape of latent kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 2010, 6, e1000935. [Google Scholar] [CrossRef] [PubMed]
- Mercier, A.; Arias, C.; Madrid, A.S.; Holdorf, M.M.; Ganem, D. Site-specific association with host and viral chromatin by kaposi’s sarcoma-associated herpesvirus LANA and its reversal during lytic reactivation. J. Virol. 2014, 88, 6762–6777. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Tsai, K.; Chen, H.S.; Wikramasinghe, P.; Davuluri, R.V.; Showe, L.; Domsic, J.; Marmorstein, R.; Lieberman, P.M. Identification of host-chromosome binding sites and candidate gene targets for kaposi’s sarcoma-associated herpesvirus LANA. J. Virol. 2012, 86, 5752–5762. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; Simon, J.M.; Lieb, J.D.; Davis, I.J.; Damania, B.; Dittmer, D.P. The open chromatin landscape of kaposi’s sarcoma-associated herpesvirus. J. Virol. 2013, 87, 11831–11842. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.L. Methylated cytosine and the brain: A new base for neuroscience. Neuron 2001, 30, 649–652. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Newell-Price, J.; Clark, A.J.; King, P. DNA methylation and silencing of gene expression. Trends Endocrinol. Metab. 2000, 11, 142–148. [Google Scholar] [CrossRef]
- Shull, A.; Noonepalle, S.; Lee, E.-J.; Choi, J.-H.; Shi, H. Sequencing the cancer methylome. In Cancer Epigenetics; Verma, M., Ed.; Springer: New York, NY, USA, 2015; pp. 627–651. [Google Scholar]
- Gavrilov, A.; Eivazova, E.; Priozhkova, I.; Lipinski, M.; Razin, S.; Vassetzky, Y. Chromosome conformation capture (from 3C to 5C) and its chip-based modification. Methods Mol. Biol. 2009, 567, 171–188. [Google Scholar] [PubMed]
- Van Berkum, N.L.; Dekker, J. Determining spatial chromatin organization of large genomic regions using 5C technology. Methods Mol. Biol. 2009, 567, 189–213. [Google Scholar] [PubMed]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. Faire (formaldehyde-assisted isolation of regulatory elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.A.; Liu, X.S. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat. Rev. Genet. 2014, 15, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Jung, J.U. The chromatin landscape of kaposi’s sarcoma-associated herpesvirus. Viruses 2013, 5, 1346–1373. [Google Scholar] [CrossRef] [PubMed]
- Uppal, T.; Jha, H.C.; Verma, S.C.; Robertson, E.S. Chromatinization of the KSHV genome during the KSHV life cycle. Cancers 2015, 7, 112–142. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Lee, H.R.; Izumiya, Y.; Tepper, C.; Kung, H.J.; Jung, J.U. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog. 2013, 9, e1003813. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; Lu, J.; Verma, S.C.; Banerjee, S.; Mehta, D.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus genome programming during the early stages of primary infection of peripheral blood mononuclear cells. MBio 2014. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ueda, K.; Sakakibara, S.; Okuno, T.; Parravicini, C.; Corbellino, M.; Yamanishi, K. Activation of latent kaposi’s sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc. Natl. Acad. Sci. USA 2001, 98, 4119–4124. [Google Scholar] [CrossRef] [PubMed]
- Uppal, T.; Banerjee, S.; Sun, Z.; Verma, S.C.; Robertson, E.S. KSHV LANA—The master regulator of KSHV latency. Viruses 2014, 6, 4961–4998. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Kaye, K.M. The latency-associated nuclear antigen, a multifunctional protein central to kaposi’s sarcoma-associated herpesvirus latency. Future Microbiol. 2011, 6, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Lan, K.; Robertson, E. Structure and function of latency-associated nuclear antigen. Curr. Top. Microbiol. Immunol. 2007, 312, 101–136. [Google Scholar] [PubMed]
- Garber, A.C.; Shu, M.A.; Hu, J.; Renne, R. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of kaposi’s sarcoma-associated herpesvirus. J. Virol. 2001, 75, 7882–7892. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Hu, J.; Pierce, B.; Weng, Z.; Renne, R. Mutational analysis of the latency-associated nuclear antigen DNA-binding domain of kaposi’s sarcoma-associated herpesvirus reveals structural conservation among gammaherpesvirus origin-binding proteins. J. Gen. Virol. 2010, 91, 2203–2215. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yang, Y.; Turner, P.C.; Jain, V.; McIntyre, L.M.; Renne, R. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog. 2014, 10, e1004240. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Naranatt, P.P.; Smith, M.S.; Zeng, L.; Bloomer, C.; Chandran, B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 2004, 78, 3601–3620. [Google Scholar] [PubMed]
- Lan, K.; Kuppers, D.A.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein jkappa, the major downstream effector of the notch signaling pathway. J. Virol. 2005, 79, 3468–3478. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Verma, S.C.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: A potential mechanism for virus-mediated control of latency. J. Virol. 2004, 78, 6585–6594. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Fitzgerald, L.D.; van Geelen, A.; Izumiya, Y.; Ellison, T.J.; Wang, D.H.; Ann, D.K.; Luciw, P.A.; Kung, H.J. Kruppel-associated box domain-associated protein-1 as a latency regulator for kaposi’s sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res. 2009, 69, 5681–5689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, C.; Guo, Y.; Wei, F.; Lu, J.; Qin, J.; Banerjee, S.; Wang, J.; Shang, H.; Verma, S.C.; et al. Inhibition of KAP1 enhances hypoxia-induced kaposi’s sarcoma-associated herpesvirus reactivation through RBP-Jκ. J. Virol. 2014, 88, 6873–6884. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Black, J.B.; Goldsmith, C.S.; Browning, P.J.; Bhalla, K.; Offermann, M.K. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J. Gen. Virol. 1999, 80, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Townes, T.M. Molecular mechanism for silencing virally transduced genes involves histone deacetylation and chromatin condensation. Proc. Natl. Acad. Sci. USA 2000, 97, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Bowser, B.S.; DeWire, S.M.; Damania, B. Transcriptional regulation of the K1 gene product of kaposi’s sarcoma-associated herpesvirus. J. Virol. 2002, 76, 12574–12583. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Song, M.J.; Chu, J.T.; Sun, R. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (kaposi’s sarcoma-associated herpesvirus). J. Virol. 2002, 76, 8252–8264. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Shedd, D.; Miller, G. Two subclasses of kaposi’s sarcoma-associated herpesvirus lytic cycle promoters distinguished by open reading frame 50 mutant proteins that are deficient in binding to DNA. J. Virol. 2005, 79, 8750–8763. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ye, F.; Xie, J.; Kuhne, K.; Gao, S.J. Genome-wide identification of binding sites for kaposi’s sarcoma-associated herpesvirus lytic switch protein, Rta. Virology 2009, 386, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Ziegelbauer, J.; Grundhoff, A.; Ganem, D. Exploring the DNA binding interactions of the kaposi’s sarcoma-associated herpesvirus lytic switch protein by selective amplification of bound sequences in vitro. J. Virol. 2006, 80, 2958–2967. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.R.; Wong, L.Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010, 6, e1001013. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G.S. Kaposi’s sarcoma-associated herpesvirus noncoding polyadenylated nuclear RNA interacts with virus- and host cell-encoded proteins and suppresses expression of genes involved in immune modulation. J. Virol. 2011, 85, 13290–13297. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Wiedmer, A.; Yuan, Y.; Robertson, E.; Lieberman, P.M. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog. 2011, 7, e1002140. [Google Scholar] [CrossRef] [PubMed]
- Tempera, I.; Lieberman, P.M. Chromatin organization of gammaherpesvirus latent genomes. Biochim. Biophys. Acta 2010, 1799, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Li, D.J.; Verma, D.; Mosbruger, T.; Swaminathan, S. CTCF and RAD21 act as host cell restriction factors for kaposi’s sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLoS Pathog. 2014, 10, e1003880. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Cheng, C.Y.; Campbell, M.; Yang, Y.C.; Hsu, H.W.; Chang, T.Y.; Chu, C.H.; Lee, Y.W.; Hung, C.L.; Lai, S.M.; et al. The chromatin modification by SUMO-2/3 but not SUMO-1 prevents the epigenetic activation of key immune-related genes during kaposi’s sarcoma associated herpesvirus reactivation. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dominguez, M.; Reyes, J.C. SUMO association with repressor complexes, emerging routes for transcriptional control. Biochim. Biophys. Acta 2009, 1789, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Izumiya, Y.; Wu, C.Y.; Fitzgerald, L.D.; Campbell, M.; Ellison, T.J.; Lam, K.S.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus (KSHV) encodes a SUMO E3 ligase that is SIM-dependent and SUMO-2/3-specific. J. Biol. Chem. 2010, 285, 5266–5273. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Hsu, H.W.; Campbell, M.; Cheng, C.Y.; Chang, P.C. K-bZip mediated SUMO-2/3 specific modification on the KSHV genome negatively regulates lytic gene expression and viral reactivation. PLoS Pathog. 2015, 11, e1005051. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Strand | Start–Stop | Small Open Reading Frame (sORF) | Upstream Open Reading Frame (uORF) | New Features |
|---|---|---|---|---|---|
| ORF6.1 | + | 3027–3203 | - | uORF | - |
| ORF6.2 | + | 3150–3203 | - | uORF | - |
| ORF10.1 | + | 14,451–14,531 | - | uORF | - |
| ORF10.2 | + | 15,574–15,756 | - | - | Internal ORF |
| ORF11.1 | + | 15,633–15,722 | - | uORF | - |
| ORF11.2 | + | 15,648–15,722 | - | uORF | - |
| ORF11.3 | + | 15,693–15,722 | - | uORF | - |
| ORF11.4 | + | 15,745–15,756 | - | uORF | - |
| ORF11.5 | + | 15,926–15,991 | - | - | Internal ORF |
| vIL6.6 | − | 17,873–17,862 | - | uORF | - |
| vIL6.5 | − | 17,915–17,877 | - | uORF | - |
| vIL6.4 | − | 18,003–17,902 | - | uORF | - |
| vIL6.3 | − | 18,047–17,985 | - | uORF | - |
| vIL6.2 | − | 18,086–18,057 | - | uORF | - |
| vIL6.1 | − | 18,116–18,057 | - | uORF | - |
| ORFK3A | − | 19,128–18,589 | - | - | Internal ORF |
| ORF70A | − | 21,099–20,038 | - | - | Alternate start |
| ORFK4A | − | 21,820–21,743 | - | uORF | - |
| ORFK4.1a | − | 22,517–22,416 | sORF | - | - |
| ORFK4.1d | − | 22,610–22,545 | sORF | - | - |
| ORFK4.1e | − | 22,653–22,627 | sORF | - | - |
| ORFK4.1c | − | 22,806–22,723 | sORF | - | - |
| ORFK4.1b | − | 22,850–22,545 | sORF | - | - |
| 1.4KbB | + | 24,871–24,915 | sORF | - | - |
| 1.4KbC | + | 24,921–25,058 | sORF | - | - |
| ORFK5.1 | − | 26,569–26,555 | - | uORF | - |
| ORFK6.1 | − | 27,647–27,615 | - | uORF | - |
| ORFK6A | − | 27,443–27,087 | - | - | Alternate start |
| ORFK6B | − | 27,422–27,087 | - | - | Alternate start |
| PAN1.1 | + | 28,655–28,768 | sORF | - | - |
| PAN1.2 | + | 28,831–28,965 | sORF | - | - |
| PAN1.3 | + | 28,888–28,965 | sORF | - | - |
| ORF20A | − | 35,322–34,429 | - | - | Internal ORF |
| ORF20B | − | 35,202–34,429 | - | - | Internal ORF |
| ORF21.1 | + | 35,151–35,177 | - | uORF | - |
| ORF25.1 | + | 42,345–42,380 | - | uORF | - |
| ORF28.1 | + | 48,758–48,811 | - | uORF | - |
| ORF30.1 | + | 50,317–50,358 | - | uORF | - |
| ORF34.1 | + | 54,399–54,485 | - | uORF | - |
| ORF35.1 | + | 55,419–55,445 | - | uORF | - |
| ORF35.2 | + | 55,442–55,474 | - | uORF | - |
| ORF37.1 | + | 57,040–57,126 | - | uORF | - |
| ORF38.1 | + | 58,251–58,259 | - | uORF | - |
| ORF38.2 | + | 58,455–58,589 | - | uORF | - |
| 43.1-AS | + | 63,214–63,228 | sORFs | - | - |
| ORF43.2-AS | + | 63,254–63,295 | sORFs | - | - |
| ORF45.1 | − | 68,447–68,364 | - | uORF | - |
| ORF49.1 | − | 72,425–72,384 | - | uORF | - |
| ORF50AS | − | 74,222–74,130 | sORF | - | - |
| k8.1 short | + | 75,890–75,971 | - | - | Internal ORF |
| ORF54A | + | 77,552–78,439 | - | - | Alternate start |
| ORF55.1 | − | 79,501–79,340 | - | uORF | - |
| ORF57A | + | 81,464–83,453 | - | - | Splice variant |
| ORF61.2 | − | 100,071–100,018 | - | uORF | - |
| ORF61.1 | − | 100,095–100,018 | - | uORF | - |
| ORF62B | − | 101,019–100,018 | - | - | Alternate start |
| ORF62A | − | 101,061–100,018 | - | - | Alternate start |
| ORF65.1 | − | 112,321–112,289 | - | uORF | - |
| ORF69.1 | + | 116,043–116,138 | - | uORF | - |
| Kaposin C2 | − | 119,084–117,738 | - | - | Alternate start |
| ORF72.1 | − | 124,182–124,108 | - | uORF | - |
| ORF75.1 | − | 134,809–134,729 | - | uORF | - |
| ORF75.2 | − | 134,894–134,817 | - | uORF | - |
| K15.1 | − | 135,938–135,846 | sORF | - | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strahan, R.; Uppal, T.; Verma, S.C. Next-Generation Sequencing in the Understanding of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Biology. Viruses 2016, 8, 92. https://doi.org/10.3390/v8040092
Strahan R, Uppal T, Verma SC. Next-Generation Sequencing in the Understanding of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Biology. Viruses. 2016; 8(4):92. https://doi.org/10.3390/v8040092
Chicago/Turabian StyleStrahan, Roxanne, Timsy Uppal, and Subhash C. Verma. 2016. "Next-Generation Sequencing in the Understanding of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Biology" Viruses 8, no. 4: 92. https://doi.org/10.3390/v8040092
APA StyleStrahan, R., Uppal, T., & Verma, S. C. (2016). Next-Generation Sequencing in the Understanding of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Biology. Viruses, 8(4), 92. https://doi.org/10.3390/v8040092