
Protein 2B of Coxsackievirus B3 Induces Autophagy Relying on Its Transmembrane Hydrophobic Sequences



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Chemicals
2.2. Cell Culture
2.3. Virus Infection
2.4. Plasmid Construction
2.5. Transfection
2.6. Western Blot
3. Results
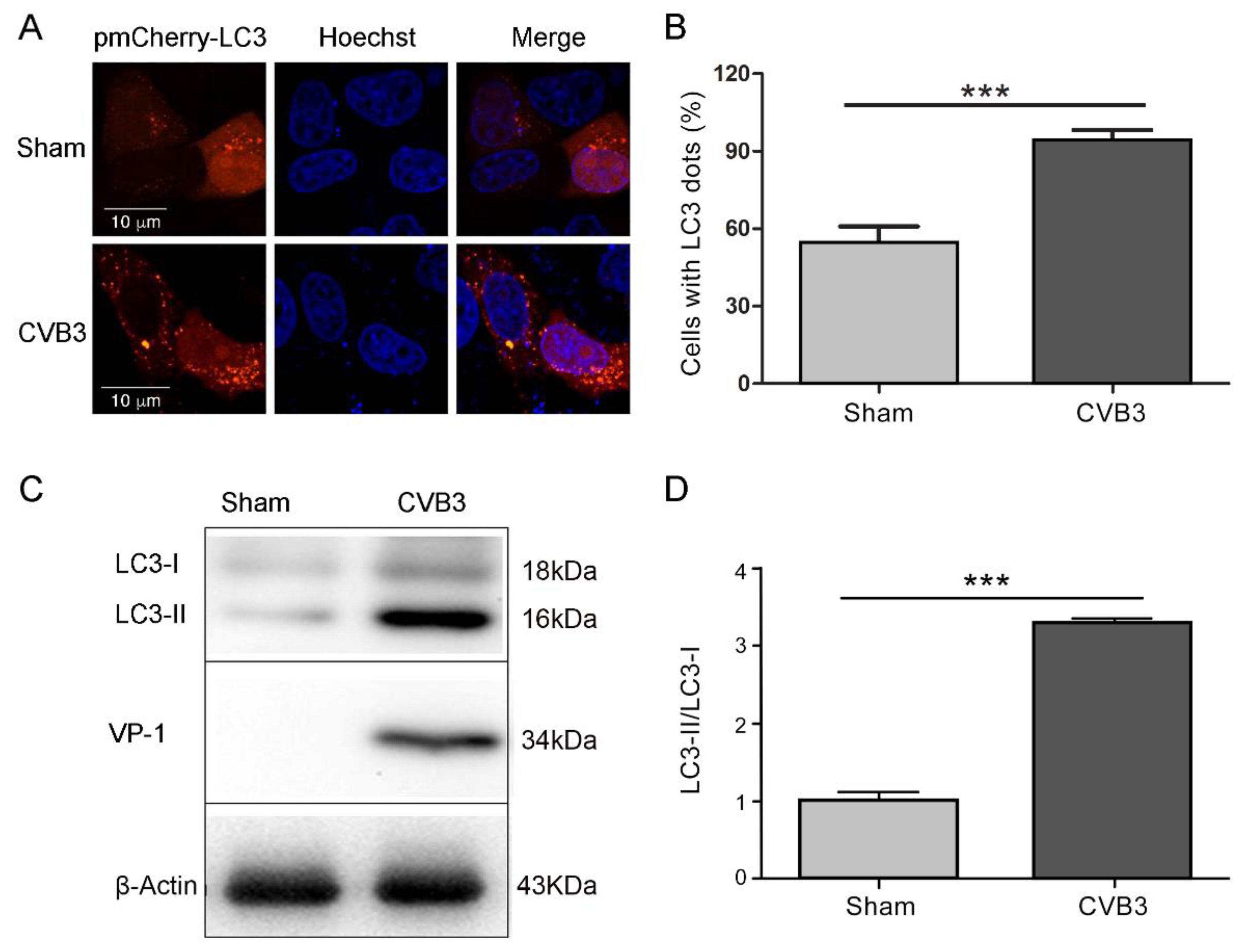
3.1. CVB3 Replication Induces Autophagy in HeLa Cells
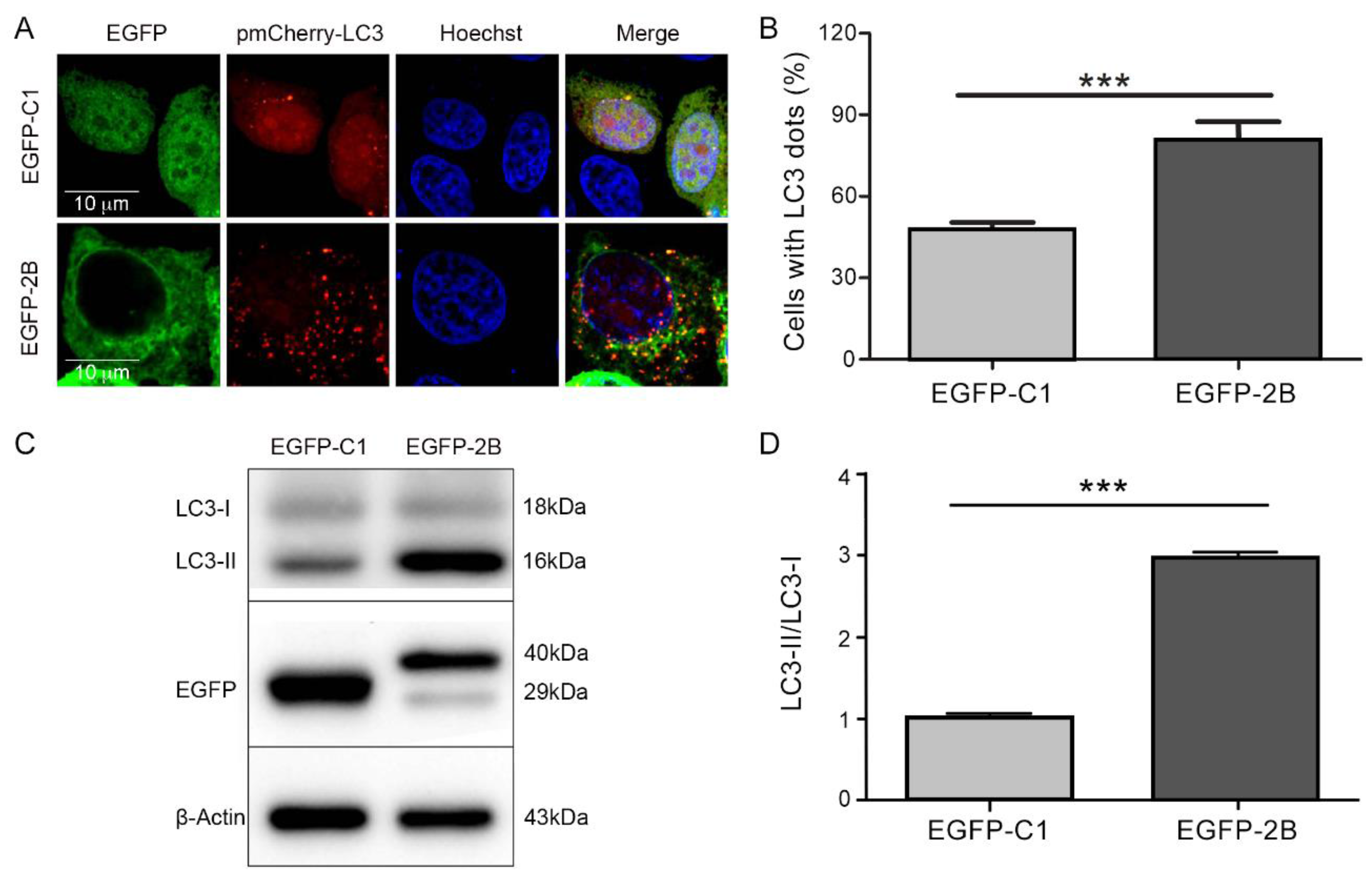
3.2. Protein 2B of CVB3 Induces Autophagy
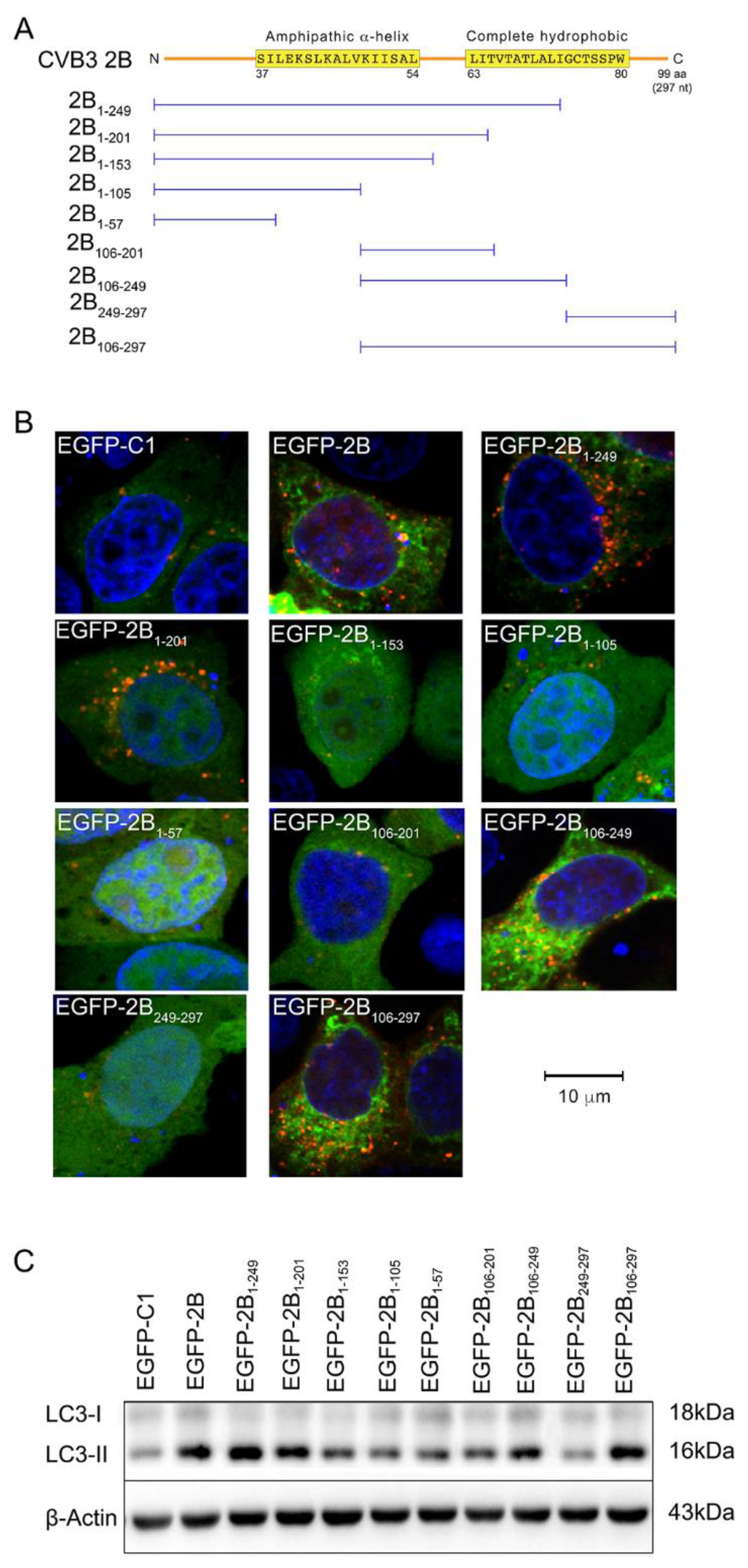
3.3. The Autophagy-Inducing Motif of Protein 2B
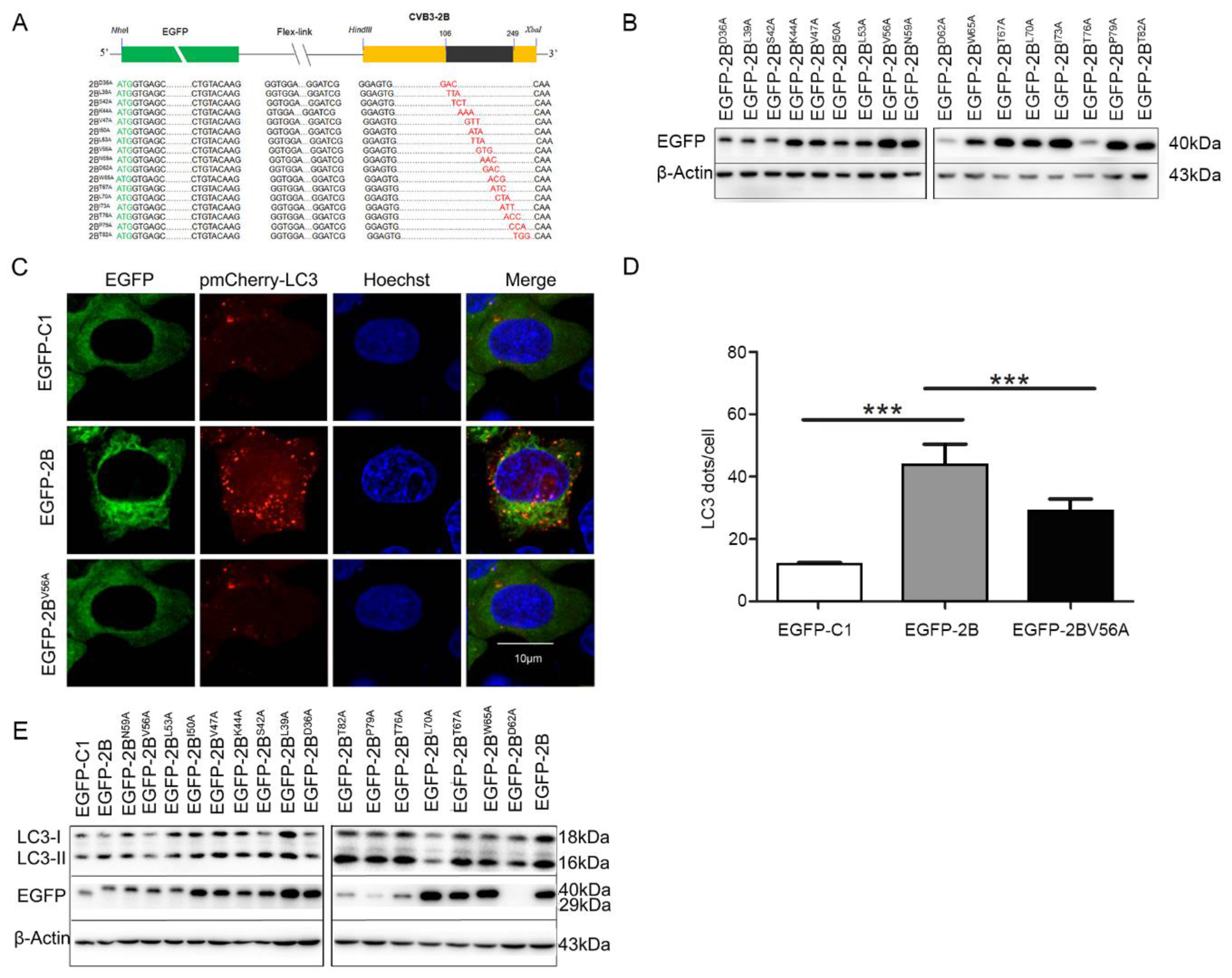
3.4. 56 Valine Residue in between the Transmembrane Sequences of Protein 2B is Essential for Autophagy-Induction
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CVB | coxsackievirus B |
| UTR | untranslated region |
| ORF | open reading frame |
| MOI | multiplicity of infection |
| pfu | plaque-forming unit |
| EGFP | enhanced green fluorescence protein |
| LC3 | microtubule-associated protein 1light chain 3 |
| PCR | polymerase chain reaction |
References
- Garmaroudi, F.S.; Marchant, D.; Hendry, R.; Luo, H.; Yang, D.; Ye, X.; Shi, J.; McManus, B.M.; et al. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. 2015, 10, 629–653. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, X.; Yu, Y.; Yu, Y.; Xie, Y.; Zou, Y.; Ge, J.; Peng, T.; Chen, R. Coxsackievirus B3-induced calpain activation facilitates the progeny virus replication via a likely mechanism related with both autophagy enhancement and apoptosis inhibition in the early phase of infection: An in vitro study in H9c2 cells. Virus Res. 2014, 179, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiao, Z.; He, F.; Zou, J.; Wu, S.; Liu, Z. MicroRNAs regulate the pathogenesis of CVB3-induced viral myocarditis. Intervirology 2013, 56, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Steinke, K.; Sachse, F.; Ettischer, N.; Strutz-Seebohm, N.; Henrion, U.; Rohrbeck, M.; Klosowski, R.; Wolters, D.; Brunner, S.; Franz, W.M.; et al. Coxsackievirus B3 modulates cardiac ion channels. FASEB J. 2013, 27, 4108–4121. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Huang, Y.C.; Yang, S.; Tsao, K.C.; Chen, C.J.; Hsieh, Y.C.; Chiu, C.H.; Lin, T.Y. Clinical features of coxsackievirus A4, B3 and B4 infections in children. PLoS ONE 2014, 9, e87391. [Google Scholar] [CrossRef] [PubMed]
- Esfandiarei, M.; McManus, B.M. Molecular biology and pathogenesis of viral myocarditis. Annu. Rev. Pathol. 2008, 3, 127–155. [Google Scholar] [CrossRef]
- Gaaloul, I.; Riabi, S.; Harrath, R.; Evans, M.; Salem, N.H.; Mlayeh, S.; Huber, S.; Aouni, M. Sudden unexpected death related to enterovirus myocarditis: Histopathology, immunohistochemistry and molecular pathology diagnosis at post-mortem. BMC Infect. Dis. 2012, 12, 212. [Google Scholar] [CrossRef] [PubMed]
- Spanakis, N.; Manolis, E.N.; Tsakris, A.; Tsiodras, S.; Panagiotopoulos, T.; Saroglou, G.; Legakis, N.J. Coxsackievirus B3 sequences in the myocardium of fatal cases in a cluster of acute myocarditis in Greece. J. Clin. Pathol. 2005, 58, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lou, C.; Liu, P. Interleukin-27 ameliorates coxsackievirus-B3-induced viral myocarditis by inhibiting Th17 cells. Virol. J. 2015, 12, 189. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Yue, Y.; Dong, N.; Xiong, S. Endoplasmic Reticulum Stress Aggravates Viral Myocarditis by Raising Inflammation through the IRE1-Associated NF-kappaB Pathway. Can. J. Cardiol. 2015, 31, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, X.; Zhang, J.; Huang, C. Antiviral and myocyte protective effects of IL-28A in coxsackievirus B3-induced myocarditis. Braz. J. Infect. Dis. 2015, 19, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Cen, Z.; Guo, Y.; Kong, Q.; Zhou, Q.; Wu, W. IL-10-producing B cells involved in the pathogenesis of Coxsackie virus B3-induced acute viral myocarditis. Int. J. Clin. Exp. Pathol. 2015, 8, 830–835. [Google Scholar] [PubMed]
- Zhao, Z.; Cai, T.Z.; Lu, Y.; Liu, W.J.; Cheng, M.L.; Ji, Y.Q. Coxsackievirus B3 induces viral myocarditis by upregulating toll-like receptor 4 expression. Biochemistry (Mosc.) 2015, 80, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Kim, K.S. Persistent coxsackievirus infection: Enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr. Top. Microbiol. Immunol. 2008, 323, 275–292. [Google Scholar] [PubMed]
- Tong, L.; Lin, L.; Wu, S.; Guo, Z.; Wang, T.; Qin, Y.; Wang, R.; Zhong, X.; Wu, X.; Wang, Y.; et al. MiR-10a* up-regulates coxsackievirus B3 biosynthesis by targeting the 3D-coding sequence. Nucleic Acids Res. 2013, 41, 3760–3771. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Zhang, Y.; He, F.; Wang, C.; Xiao, Z.; Zou, J.; Wang, F.; Liu, Z. Short hairpin RNA targeting 2B gene of coxsackievirus B3 exhibits potential antiviral effects both in vitro and in vivo. BMC Infect. Dis. 2012, 12, 177. [Google Scholar] [CrossRef] [PubMed]
- Patargias, G.; Barke, T.; Watts, A.; Fischer, W.B. Model generation of viral channel forming 2B protein bundles from polio and coxsackie viruses. Mol. Membr. Biol. 2009, 26, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Sanchez-Martinez, S.; Carrasco, L.; Nieva, J.L. A peptide based on the pore-forming domain of pro-apoptotic poliovirus 2B viroporin targets mitochondria. Biochim. Biophys. Acta 2010, 1798, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Van Kuppeveld, F.J.; Galama, J.M.; Zoll, J.; van den Hurk, P.J.; Melchers, W.J. Coxsackie B3 virus protein 2B contains cationic amphipathic helix that is required for viral RNA replication. J. Virol. 1996, 70, 3876–3886. [Google Scholar]
- Van Kuppeveld, F.J.; Hoenderop, J.G.; Smeets, R.L.; Willems, P.H.; Dijkman, H.B.; Galama, J.M.; Melchers, W.J. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 1997, 16, 3519–3532. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.S.; de Mattia, F.; Van Dommelen, M.M.; Lanke, K.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. Functional analysis of picornavirus 2B proteins: Effects on calcium homeostasis and intracellular protein trafficking. J. Virol. 2008, 82, 3782–3790. [Google Scholar] [CrossRef] [PubMed]
- Campanella, M.; de Jong, A.S.; Lanke, K.W.; Melchers, W.J.; Willems, P.H.; Pinton, P.; Rizzuto, R.; van Kuppeveld, F.J. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 2004, 279, 18440–18450. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Luo, H. Interplay between the cellular autophagy machinery and positive-stranded RNA viruses. Acta Biochim. Biophys. Sin. (Shanghai) 2012, 44, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Kemball, C.C.; Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Harkins, S.; Kiosses, W.B.; Whitton, J.L. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 2010, 84, 12110–12124. [Google Scholar] [CrossRef] [PubMed]
- Mandelbaum, J.; Rollins, N.; Shah, P.; Bowman, D.; Lee, J.Y.; Tayber, O.; Bernard, H.; LeRoy, P.; Li, P.; Koenig, E.; et al. Identification of a lung cancer cell line deficient in atg7-dependent autophagy. Autophagy 2015. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.H.; Lian, S.; Zhao, R.; Rao, X.M.; McMasters, K.M.; Zhou, H.S. Combination of autophagy inducer rapamycin and oncolytic adenovirus improves antitumor effect in cancer cells. Virol. J. 2013, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Kyles, P.; Tyagi, S.C.; Tyagi, N. Autophagy of mitochondria: A promising therapeutic target for neurodegenerative disease. Cell Biochem. Biophys. 2014, 70, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Bai, B.; Yu, B.; Wang, T.; Wang, H.; Wang, Y.; Li, H.; Tong, L.; Wang, Y.; Zhang, F.; et al. Coxsackievirus B3 Induces Autophagic Response in Cardiac Myocytes in vivo. Biochemistry (Mosc.) 2015, 80, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yu, B.; Lin, L.; Zhai, X.; Han, Y.; Qin, Y.; Guo, Z.; Wu, S.; Zhong, X.; Wang, Y.; et al. A functional nuclear localization sequence in the VP1 capsid protein of coxsackievirus B3. Virology 2012, 433, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Wileman, T. Aggresomes and autophagy generate sites for virus replication. Science 2006, 312, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Chau, D.H.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2007, 12, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.J.; Ye, X.; Qiu, Y.; Zhang, H.M.; Hemida, M.G.; Wang, F.; Lim, T.; Gu, A.; Cho, B.; Kim, H.; et al. Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES-containing genes. Cell Death Differ. 2015. [Google Scholar] [CrossRef] [PubMed]
- Moffat, K.; Howell, G.; Knox, C.; Belsham, G.J.; Monaghan, P.; Ryan, M.D.; Wileman, T. Effects of foot-and-mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum-to-Golgi transport. J. Virol. 2005, 79, 438243–438295. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.S.; Wessels, E.; Dijkman, H.B.; Galama, J.M.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. Determinants for membrane association and permeabilization of the coxsackievirus 2B protein and the identification of the Golgi complex as the target organelle. J. Biol. Chem. 2003, 278, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.S.; Melchers, W.J.; Glaudemans, D.H.; Willems, P.H.; van Kuppeveld, F.J. Mutational analysis of different regions in the coxsackievirus 2B protein: Requirements for homo-multimerization, membrane permeabilization, subcellular localization, and virus replication. J. Biol. Chem. 2004, 279, 19924–19935. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martinez, S.; Madan, V.; Carrasco, L.; Nieva, J.L. Membrane-active peptides derived from picornavirus 2B viroporin. Curr. Protein Pept. Sci. 2012, 13, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Sanchez-Martinez, S.; Vedovato, N.; Rispoli, G.; Carrasco, L.; Nieva, J.L. Plasma membrane-porating domain in poliovirus 2B protein. A short peptide mimics viroporin activity. J. Mol. Biol. 2007, 374, 951–964. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.; Zhai, X.; Chen, Y.; Wang, R.; Lin, L.; Chen, S.; Wang, T.; Zhong, X.; Wu, X.; Wang, Y.; et al. Protein 2B of Coxsackievirus B3 Induces Autophagy Relying on Its Transmembrane Hydrophobic Sequences. Viruses 2016, 8, 131. https://doi.org/10.3390/v8050131
Wu H, Zhai X, Chen Y, Wang R, Lin L, Chen S, Wang T, Zhong X, Wu X, Wang Y, et al. Protein 2B of Coxsackievirus B3 Induces Autophagy Relying on Its Transmembrane Hydrophobic Sequences. Viruses. 2016; 8(5):131. https://doi.org/10.3390/v8050131
Chicago/Turabian StyleWu, Heng, Xia Zhai, Yang Chen, Ruixue Wang, Lexun Lin, Sijia Chen, Tianying Wang, Xiaoyan Zhong, Xiaoyu Wu, Yan Wang, and et al. 2016. "Protein 2B of Coxsackievirus B3 Induces Autophagy Relying on Its Transmembrane Hydrophobic Sequences" Viruses 8, no. 5: 131. https://doi.org/10.3390/v8050131
APA StyleWu, H., Zhai, X., Chen, Y., Wang, R., Lin, L., Chen, S., Wang, T., Zhong, X., Wu, X., Wang, Y., Zhang, F., Zhao, W., & Zhong, Z. (2016). Protein 2B of Coxsackievirus B3 Induces Autophagy Relying on Its Transmembrane Hydrophobic Sequences. Viruses, 8(5), 131. https://doi.org/10.3390/v8050131