
Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
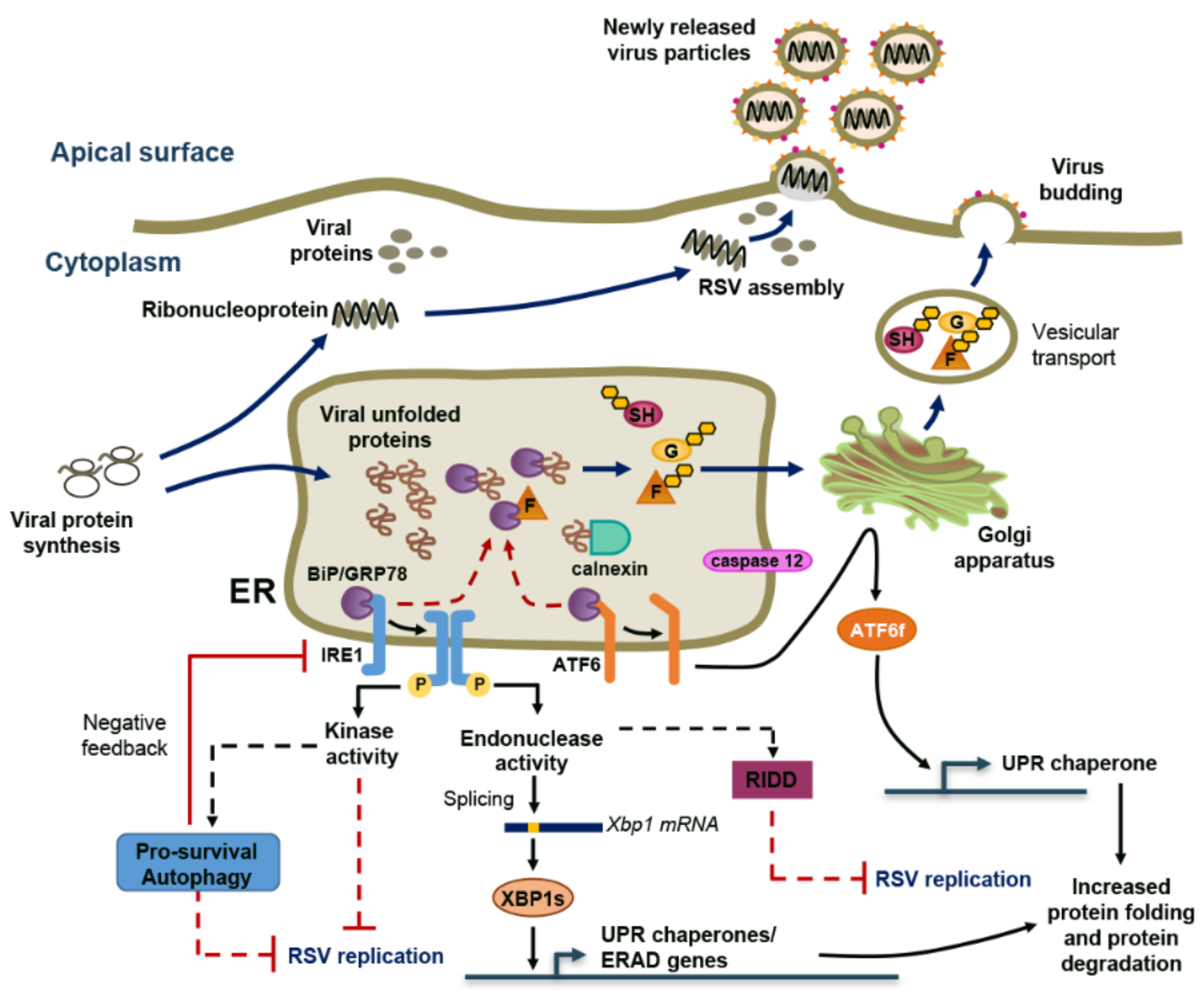
2. RSV Induces ER Stress and a Non-Canonical Unfolded-Protein Response (UPR)
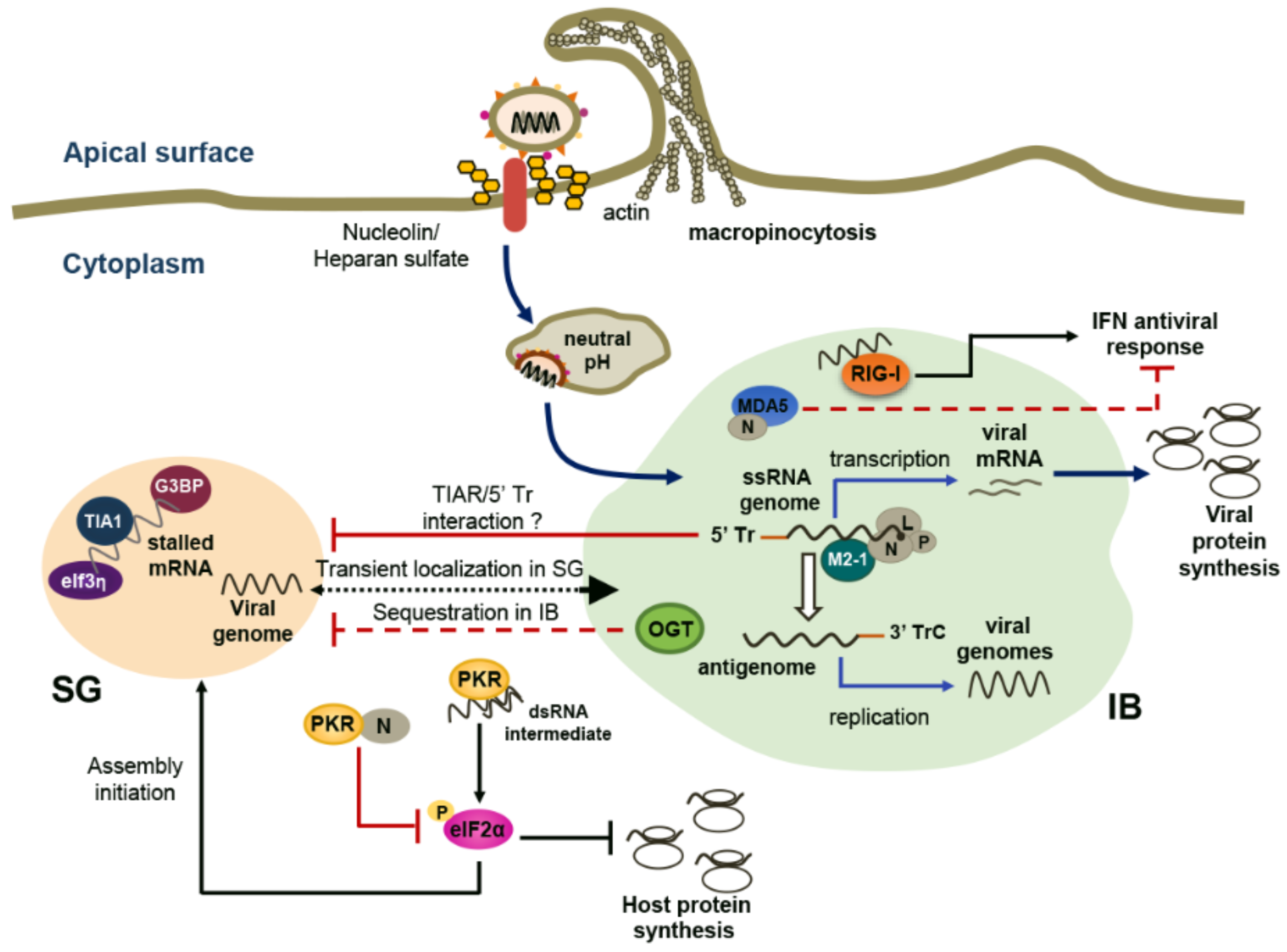
3. RSV Takes Advantage of Cytoplasmic SGs and Specific IBs
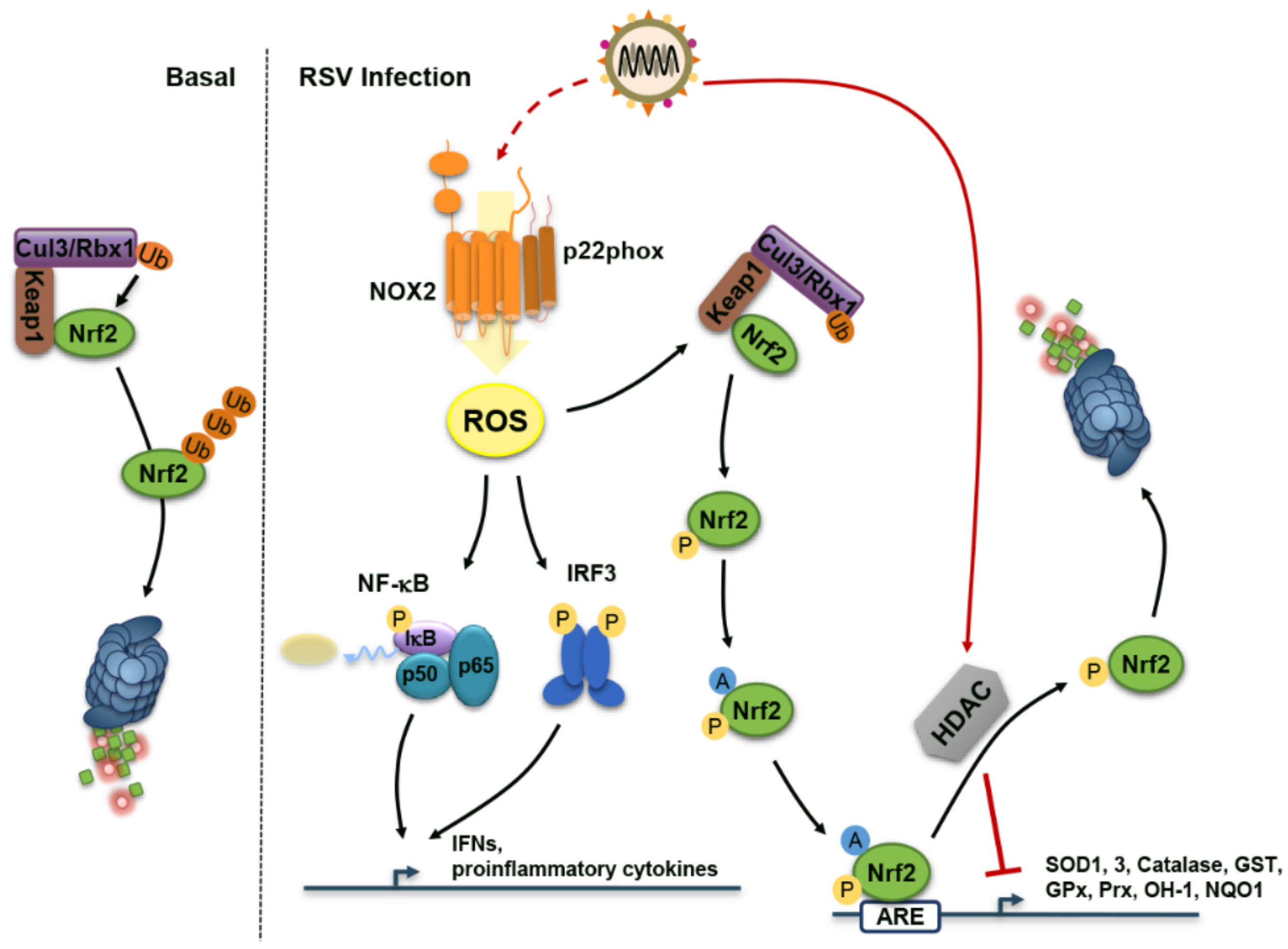
4. Reactive Oxygen Species (ROS) During RSV Infection: Too Much of a Good Thing
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AEC | Airway epithelial cell |
| AOE | Antioxidant enzyme |
| AP-1 | Activator protein-1 |
| ARE | Antioxidant response element |
| ATF6 | Activated transcription factor 6 |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| BAL | Bronchoalveolar lavage |
| BHA | Butylated hydroxyanisol |
| BiP/GRP78 | Binding immunoglobulin protein |
| Cul3 | Cullin 3 |
| DBSA | 3,5-dibromosalicylaldehide |
| DC | Dendritic cell |
| dsRNA | Double-stranded RNA |
| DUOX2 | Dual oxidase 2 |
| eIF | Eukaryotic translation initiation factor |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated protein degradation |
| G3BP1 | RasGAP SH3 domain-binding protein 1 |
| GPx | Glutathione peroxidase |
| GSH | Glutathione |
| GSSG | Oxidized glutathione |
| GST | Glutathione S-transferase |
| HDAC | Histone deacetylase |
| HeV | Hendra virus |
| HN | Hemagglutinin-neuraminidase |
| HNE | 4-hydroxynonenal |
| HTBE | Human tracheobronchial epithelial |
| IB | Inclusion body |
| IFN | Interferon |
| IRE1 | Inositol-requiring enzyme 1 |
| IRF | Interferon regulatory factor |
| JNK | c-Jun N-terminal kinase |
| Keap1 | Kelch-like ECH-associated protein 1 |
| KO | Knockout |
| L | Large polymerase protein |
| LC3b | Microtubule-associated proteins 1A/1B light chain 3B |
| Le | Leader |
| MAPK | Mitogen-activated protein kinase |
| MAVS | Mitochondrial antiviral-signaling Protein |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MDA | Malondialdehyde |
| MDA5 | Melanoma differentiation-associated protein 5 |
| MEFs | Mouse embryonic fibroblasts |
| MPV | Metapneumovirus |
| MuV | Mumps virus |
| MV | Measles virus |
| NiV | Nipah virus |
| NAC | N-acetylcysteine |
| NDV | Newcastle disease virus |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NHBE | Normal human bronchial epithelial cell |
| NLRP3 | Nucleotide-binding domain and leucine-rich repeat, pyrin domain containing 3 |
| NOX2 | NADPH oxidase 2 |
| NPS | Nasopharyngeal Secretion |
| NQO1 | NAD(P)H:quinone oxidoreductase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NS | Non-structural |
| OGN | O-linked N-acetylglucosamine |
| OGT | OGN transferase |
| OH-1 | Heme oxygenase 1 |
| PAMP | Pathogen-associated molecular pattern |
| PERK | PKR-like ER kinase |
| PIV | Parainfluenza viruses |
| PKR | Protein kinase R |
| RNP | RNA-binding protein |
| PRR | Pattern recognition receptor |
| Prx | Peroxiredoxin |
| Rbx1 | Ring-box 1 |
| RIDD | IRE1-dependent decay |
| RIG-I | Retinoic acid-inducible gene I |
| RLR | RIG-I-like receptor |
| ROS | Reactive oxygen species |
| RSV | Respiratory syncytial virus |
| SAE | Small alveolar epithelial cell |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| SeV | Sendai virus |
| SG | Stress granule |
| SOD | Superoxide dismutase |
| Th2 | T helper-type 2 |
| TIA1 | T-cell intracellular antigen |
| TIAR | TIA1-related |
| TLR | Toll-like receptor |
| TNFα | Tumor necrosis factor alpha |
| Tr | Trailer sequence |
| TRAF2 | TNF receptor-associated factor 2 |
| TrC | Complement of the Tr sequence |
| TSA | Trichostatin A |
| UPR | Unfolded-protein response |
| Wt | wild-type |
| XBP1 | X-box binding protein 1 |
References
- Russell, C.J.; Hurwitz, J.L. Sendai virus as a backbone for vaccines against rsv and other human paramyxoviruses. Expert Rev. Vaccines 2016, 15, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Palgen, J.L.; Jurgens, E.M.; Moscona, A.; Porotto, M.; Palermo, L.M. Unity in diversity: Shared mechanism of entry among paramyxoviruses. Prog. Mol. Biol. Transl. Sci. 2015, 129, 1–32. [Google Scholar] [PubMed]
- Collins, P.L.; Melero, J.A. Progress in understanding and controlling respiratory syncytial virus: Still crazy after all these years. Virus Res. 2011, 162, 80–99. [Google Scholar] [CrossRef] [PubMed]
- Pickles, R.J.; DeVincenzo, J.P. Respiratory syncytial virus (rsv) and its propensity for causing bronchiolitis. J. Pathol. 2015, 235, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Krilov, L.R. Respiratory syncytial virus disease: Update on treatment and prevention. Expert Rev. Anti Infect. Ther. 2011, 9, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Lay, M.K.; Gonzalez, P.A.; Leon, M.A.; Cespedes, P.F.; Bueno, S.M.; Riedel, C.A.; Kalergis, A.M. Advances in understanding respiratory syncytial virus infection in airway epithelial cells and consequential effects on the immune response. Microbes Infect. 2013, 15, 230–242. [Google Scholar] [CrossRef] [PubMed]
- De Graaff, P.M.; de Jong, E.C.; van Capel, T.M.; van Dijk, M.E.; Roholl, P.J.; Boes, J.; Luytjes, W.; Kimpen, J.L.; van Bleek, G.M. Respiratory syncytial virus infection of monocyte-derived dendritic cells decreases their capacity to activate cd4 t cells. J. Immunol. 2005, 175, 5904–5911. [Google Scholar] [CrossRef] [PubMed]
- Dakhama, A.; Kaan, P.M.; Hegele, R.G. Permissiveness of guinea pig alveolar macrophage subpopulations to acute respiratory syncytial virus infection in vitro. Chest 1998, 114, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Plata, A.; Casola, A.; Suarez, G.; Yu, X.; Spetch, L.; Peeples, M.E.; Garofalo, R.P. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2006, 34, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.R.; Johnson, C.N.; Corbett, K.S.; Edwards, G.C.; Graham, B.S. Primary human mdc1, mdc2, and pdc dendritic cells are differentially infected and activated by respiratory syncytial virus. PLoS ONE 2011, 6, e16458. [Google Scholar] [CrossRef] [PubMed]
- Noton, S.L.; Fearns, R. Initiation and regulation of paramyxovirus transcription and replication. Virology 2015, 479–480, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Parks, G.D. Paramyxoviridae: The Viruses and Their Replication, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1449–1496. [Google Scholar]
- Tayyari, F.; Marchant, D.; Moraes, T.J.; Duan, W.; Mastrangelo, P.; Hegele, R.G. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat. Med. 2011, 17, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Krzyzaniak, M.A.; Zumstein, M.T.; Gerez, J.A.; Picotti, P.; Helenius, A. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the f protein. PLoS Pathog. 2013, 9, e1003309. [Google Scholar] [CrossRef] [PubMed]
- Marr, N.; Turvey, S.E.; Grandvaux, N. Pathogen recognition receptor crosstalk in respiratory syncytial virus sensing: A host and cell type perspective. Trends Microbiol. 2013, 21, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mathews, M.B.; Mohr, I. Tinkering with translation: Protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 2013, 5, a012351. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.; Peeples, M.; Hamilton, R. Effect of respiratory syncytial virus infection of hela-cell macromolecular synthesis. J. Gen. Virol. 1977, 37, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. Respiratory syncytial virus mechanisms to interfere with type 1 interferons. Curr. Top. Microbiol. Immunol. 2013, 372, 173–191. [Google Scholar] [PubMed]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.; Gou, Y.; Zook, A.; Lozano, M.M.; Dudley, J.P. Erad and how viruses exploit it. Front. Microbiol. 2014, 5, 330. [Google Scholar] [CrossRef] [PubMed]
- Jheng, J.R.; Ho, J.Y.; Horng, J.T. Er stress, autophagy, and rna viruses. Front. Microbiol. 2014, 5, 388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, A. Virus-induced er stress and the unfolded protein response. Front. Plant Sci. 2012, 3, 293. [Google Scholar] [CrossRef] [PubMed]
- Bitko, V.; Barik, S. An endoplasmic reticulum-specific stress-activated caspase (caspase-12) is implicated in the apoptosis of a549 epithelial cells by respiratory syncytial virus. J. Cell. Biochem. 2001, 80, 441–454. [Google Scholar] [CrossRef]
- Hassan, I.; Gaines, K.S.; Hottel, W.J.; Wishy, R.M.; Miller, S.E.; Powers, L.S.; Rutkowski, D.T.; Monick, M.M. Inositol-requiring enzyme 1 inhibits respiratory syncytial virus replication. J. Biol. Chem. 2014, 289, 7537–7546. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Bartenschlager, R. Dengue virus- and hepatitis c virus-induced replication and assembly compartments: The enemy inside—Caught in the web. J. Virol. 2014, 88, 5907–5911. [Google Scholar] [CrossRef] [PubMed]
- Vasallo, C.; Gastaminza, P. Cellular stress responses in hepatitis c virus infection: Mastering a two-edged sword. Virus Res. 2015, 209, 100–117. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Garcia-Barreno, B.; Vivo, A.; Melero, J.A. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: Formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22k protein. Virology 1993, 195, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Graham, B.S. Viral and host factors in human respiratory syncytial virus pathogenesis. J. Virol. 2008, 82, 2040–2055. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Mohakud, N.K.; Pena, L.; Kumar, S. Human metapneumovirus: Review of an important respiratory pathogen. Int. J. Infect. Dis. 2014, 25, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, M.E.; Lifland, A.W.; Utley, T.J.; Santangelo, P.J.; Crowe, J.E., Jr. Respiratory syncytial virus induces host rna stress granules to facilitate viral replication. J. Virol. 2010, 84, 12274–12284. [Google Scholar] [CrossRef] [PubMed]
- He, B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Diff. 2006, 13, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L. O glycosylation of glycoprotein g of human respiratory syncytial virus is specified within the divergent ectodomain. J. Virol. 1990, 64, 4007–4012. [Google Scholar] [PubMed]
- Rixon, H.W.; Brown, G.; Aitken, J.; McDonald, T.; Graham, S.; Sugrue, R.J. The small hydrophobic (sh) protein accumulates within lipid-raft structures of the golgi complex during respiratory syncytial virus infection. J. Gen. Virol. 2004, 85, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Batonick, M.; Wertz, G.W. Requirements for human respiratory syncytial virus glycoproteins in assembly and egress from infected cells. Adv. Virol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Kwilas, S.; Liesman, R.M.; Zhang, L.; Walsh, E.; Pickles, R.J.; Peeples, M.E. Respiratory syncytial virus grown in vero cells contains a truncated attachment protein that alters its infectivity and dependence on glycosaminoglycans. J. Virol. 2009, 83, 10710–10718. [Google Scholar] [CrossRef] [PubMed]
- Brock, S.C.; Goldenring, J.R.; Crowe, J.E., Jr. Apical recycling systems regulate directional budding of respiratory syncytial virus from polarized epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 15143–15148. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.; Stott, E.J.; Wertz, G.W. Intracellular processing of the human respiratory syncytial virus fusion glycoprotein: Amino acid substitutions affecting folding, transport and cleavage. J. Gen. Virol. 1992, 73 Pt 5, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.T.; Randall, R.E.; Lamb, R.A. Intracellular maturation and transport of the sv5 type ii glycoprotein hemagglutinin-neuraminidase: Specific and transient association with grp78-bip in the endoplasmic reticulum and extensive internalization from the cell surface. J. Cell Biol. 1989, 109, 3273–3289. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Torres, J.; Liu, D.X. The emerging roles of viroporins in er stress response and autophagy induction during virus infection. Viruses 2015, 7, 2834–2857. [Google Scholar] [CrossRef] [PubMed]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The sars coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS ONE 2009, 4, e8342. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jiménez-Guardeño, J.M.; Regla-Nava, J.A.; Álvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.D.; Dent, K.C.; Atkins, E.; Foster, T.L.; Verow, M.; Gorny, P.; Harris, M.; Hiscox, J.A.; Ranson, N.A.; Griffin, S.; et al. Direct visualization of the small hydrophobic protein of human respiratory syncytial virus reveals the structural basis for membrane permeability. FEBS Lett. 2010, 584, 2786–2790. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Kar, S.; Vakakis, E.; Kotecha, S.; Triantafilou, M. Human respiratory syncytial virus viroporin sh: A viral recognition pathway used by the host to signal inflammasome activation. Thorax 2013, 68, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, S.; Tran, K.C.; Luthra, P.; Teng, M.N.; He, B. Function of the respiratory syncytial virus small hydrophobic protein. J. Virol. 2007, 81, 8361–8366. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Achazi, K.; Niedrig, M. Tick-borne encephalitis virus triggers inositol-requiring enzyme 1 (ire1) and transcription factor 6 (atf6) pathways of unfolded protein response. Virus Res. 2013, 178, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Hassan, I.H.; Zhang, M.S.; Powers, L.S.; Shao, J.Q.; Baltrusaitis, J.; Rutkowski, D.T.; Legge, K.; Monick, M.M. Influenza a viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (ire1) stress pathway. J. Biol. Chem. 2012, 287, 4679–4689. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated ire1-dependent decay of messenger rnas in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Darling, N.J.; Cook, S.J. The role of mapk signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.; Morris, S.H.; Owczarczyk, A.B.; Lukacs, N.W. Deficiency of autophagy protein map1-lc3b mediates il-17-dependent lung pathology during respiratory viral infection via er stress-associated il-1. Mucosal Immunol. 2015, 8, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Lloyd, R.E. Regulation of stress granules in virus systems. Trends Microbiol. 2012, 20, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Iseni, F.; Garcin, D.; Nishio, M.; Kedersha, N.; Anderson, P.; Kolakofsky, D. Sendai virus trailer rna binds tiar, a cellular protein involved in virus-induced apoptosis. Embo J. 2002, 21, 5141–5150. [Google Scholar] [CrossRef] [PubMed]
- Okonski, K.M.; Samuel, C.E. Stress granule formation induced by measles virus is protein kinase pkr dependent and impaired by rna adenosine deaminase adar1. J. Virol. 2013, 87, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, M.E.; Mainou, B.A.; Dermody, T.S.; Crowe, J.E., Jr. Activation of protein kinase r is required for induction of stress granules by respiratory syncytial virus but dispensable for viral replication. Virology 2011, 413, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Hanley, L.L.; McGivern, D.R.; Teng, M.N.; Djang, R.; Collins, P.L.; Fearns, R. Roles of the respiratory syncytial virus trailer region: Effects of mutations on genome production and stress granule formation. Virology 2010, 406, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Fricke, J.; Koo, L.Y.; Brown, C.R.; Collins, P.L. P38 and ogt sequestration into viral inclusion bodies in cells infected with human respiratory syncytial virus suppresses mk2 activities and stress granule assembly. J. Virol. 2013, 87, 1333–1347. [Google Scholar] [CrossRef] [PubMed]
- Groskreutz, D.J.; Babor, E.C.; Monick, M.M.; Varga, S.M.; Hunninghake, G.W. Respiratory syncytial virus limits alpha subunit of eukaryotic translation initiation factor 2 (eif2alpha) phosphorylation to maintain translation and viral replication. J. Biol. Chem. 2010, 285, 24023–24031. [Google Scholar] [CrossRef] [PubMed]
- Emara, M.M.; Brinton, M.A. Interaction of tia-1/tiar with west nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Komatsu, T.; Kitagawa, Y.; Sada, K.; Gotoh, B. Sendai virus c protein plays a role in restricting pkr activation by limiting the generation of intracellular double-stranded rna. J. Virol. 2008, 82, 10102–10110. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.; Takimoto, T. Antagonism of innate immunity by paramyxovirus accessory proteins. Viruses 2009, 1, 574–593. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Kawabata, R.; Honda, T.; Tomonaga, K.; Sakaguchi, T.; Irie, T. Ifn-beta-inducing, unusual viral rna species produced by paramyxovirus infection accumulated into distinct cytoplasmic structures in an rna-type-dependent manner. Front. Microbiol. 2015, 6, 804. [Google Scholar] [CrossRef] [PubMed]
- Santangelo, P.J.; Lifland, A.W.; Curt, P.; Sasaki, Y.; Bassell, G.J.; Lindquist, M.E.; Crowe, J.E., Jr. Single molecule-sensitive probes for imaging rna in live cells. Nat. Methods 2009, 6, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Carromeu, C.; Simabuco, F.M.; Tamura, R.E.; Farinha Arcieri, L.E.; Ventura, A.M. Intracellular localization of human respiratory syncytial virus l protein. Arch. Virol. 2007, 152, 2259–2263. [Google Scholar] [CrossRef] [PubMed]
- Ohn, T.; Kedersha, N.; Hickman, T.; Tisdale, S.; Anderson, P. A functional rnai screen links o-glcnac modification of ribosomal proteins to stress granule and processing body assembly. Nat. Cell Biol. 2008, 10, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Lifland, A.W.; Jung, J.; Alonas, E.; Zurla, C.; Crowe, J.E., Jr.; Santangelo, P.J. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by mda5 and mavs. J. Virol. 2012, 86, 8245–8258. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Mariani, M.; Fink, K. Lung epithelial nox/duox and respiratory virus infections. Clin. Sci. 2015, 128, 337–347. [Google Scholar] [CrossRef] [PubMed]
- MacNee, W. Oxidative stress and lung inflammation in airways disease. Eur. J. Pharmacol. 2001, 429, 195–207. [Google Scholar] [CrossRef]
- Chow, C.W.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative stress and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Reshi, M.L.; Su, Y.C.; Hong, J.R. Rna viruses: Ros-mediated cell death. Int. J. Cell Biol. 2014, 2014, 467452. [Google Scholar] [CrossRef] [PubMed]
- Castro, S.M.; Guerrero-Plata, A.; Suarez-Real, G.; Adegboyega, P.A.; Colasurdo, G.N.; Khan, A.M.; Garofalo, R.P.; Casola, A. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am. J. Respir. Crit. Care Med. 2006, 174, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Jantzi, P.D.; Esham, D.L.; Spratt, H.; Kurosky, A.; Casola, A.; Garofalo, R.P. Viral-mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis. Am. J. Respir. Crit. Care Med. 2011, 183, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Solis, G.; de la Torre-Aguilar, M.J.; Torres-Borrego, J.; Llorente-Cantarero, F.J.; Gil-Campos, M.; Fernandez-Gutierrez, F.; Tunez-Finana, I.; Perez-Navero, J.L. Oxidative stress and inflamatory plasma biomarkers in respiratory syncytial virus bronchiolitis. Clin. Respir. J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.P.; Hassoun, P.M.; Brower, R. Redox imbalance and ventilator-induced lung injury. Antioxid. Redox Signal. 2007, 9, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Komaravelli, N.; Mautemps, N.; Liu, T.; Garofalo, R.P.; Casola, A. Antioxidant mimetics modulate oxidative stress and cellular signaling in airway epithelial cells infected with respiratory syncytial virus. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L991–L1000. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Liu, T.; Castro, S.M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir. Cell Mol. Biol. 2009, 41, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Komaravelli, N.; Tian, B.; Ivanciuc, T.; Mautemps, N.; Brasier, A.R.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus infection down-regulates antioxidant enzyme expression by triggering deacetylation-proteasomal degradation of nrf2. Free Radic. Biol. Med. 2015, 88, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; de Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Diff. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Imani, F.; Miller-DeGraff, L.; Walters, D.; Melendi, G.A.; Yamamoto, M.; Polack, F.P.; Kleeberger, S.R. Antiviral activity of nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 2009, 179, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: Promises and perils of nrf2. Pharmacol. Rev. 2012, 64, 1055–1081. [Google Scholar] [CrossRef] [PubMed]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory syncytial virus-mediated nf-kappa b p65 phosphorylation at serine 536 is dependent on rig-i, traf6, and ikk beta. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Guan, X.; Yoboua, F.; Zucchini, N.; Fink, K.; Doyon, P.; Martin, L.; Servant, M.J.; Chartier, S. Sustained activation of interferon regulatory factor 3 during infection by paramyxoviruses requires mda5. J. Innate Immun. 2014, 6, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; et al. Distinct rig-i and mda5 signaling by rna viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic acid-inducible gene i mediates early antiviral response and toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, K.A. Oxidant stress regulation of il-8 and icam-1 gene expression: Differential activation and binding of the transcription factors ap-1 and nf-kappab (review). Int. J. Mol. Med. 1999, 4, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Mastronarde, J.G.; Monick, M.M.; Hunninghake, G.W. Oxidant tone regulates il-8 production in epithelium infected with respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 1995, 13, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Casola, A.; Burger, N.; Liu, T.; Jamaluddin, M.; Brasier, A.R.; Garofalo, R.P. Oxidant tone regulates rantes gene expression in airway epithelial cells infected with respiratory syncytial virus. Role in viral-induced interferon regulatory factor activation. J. Biol. Chem. 2001, 276, 19715–19722. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Frey, R.S.; Rahman, A.; Malik, A.B. Role of neutrophil nadph oxidase in the mechanism of tumor necrosis factor-alpha -induced nf-kappa b activation and intercellular adhesion molecule-1 expression in endothelial cells. J. Biol. Chem. 2002, 277, 3404–3411. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Castro, S.; Brasier, A.R.; Jamaluddin, M.; Garofalo, R.P.; Casola, A. Reactive oxygen species mediate virus-induced stat activation: Role of tyrosine phosphatases. J. Biol. Chem. 2004, 279, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Indukuri, H.; Castro, S.M.; Liao, S.M.; Feeney, L.A.; Dorsch, M.; Coyle, A.J.; Garofalo, R.P.; Brasier, A.R.; Casola, A. Ikkepsilon regulates viral-induced interferon regulatory factor-3 activation via a redox-sensitive pathway. Virology 2006, 353, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Duval, A.; Martel, A.; Soucy-Faulkner, A.; Grandvaux, N. Dual role of nox2 in respiratory syncytial virus- and sendai virus-induced activation of nf-kappab in airway epithelial cells. J. Immunol. 2008, 180, 6911–6922. [Google Scholar] [CrossRef] [PubMed]
- Soucy-Faulkner, A.; Mukawera, E.; Fink, K.; Martel, A.; Jouan, L.; Nzengue, Y.; Lamarre, D.; vande Velde, C.; Grandvaux, N. Requirement of nox2 and reactive oxygen species for efficient rig-i-mediated antiviral response through regulation of mavs expression. PLoS Pathog. 2010, 6, e1000930. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Martin, L.; Mukawera, E.; Chartier, S.; de Deken, X.; Brochiero, E.; Miot, F.; Grandvaux, N. Ifnbeta/tnfalpha synergism induces a non-canonical stat2/irf9-dependent pathway triggering a novel duox2 nadph oxidase-mediated airway antiviral response. Cell Res. 2013, 23, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, C.H.; Ryu, J.H.; Kim, M.J.; Park, C.Y.; Lee, J.M.; Holtzman, M.J.; Yoon, J.H. Reactive oxygen species induce antiviral innate immune response through ifn-lambda regulation in human nasal epithelial cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 855–865. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cervantes-Ortiz, S.L.; Zamorano Cuervo, N.; Grandvaux, N. Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology. Viruses 2016, 8, 124. https://doi.org/10.3390/v8050124
Cervantes-Ortiz SL, Zamorano Cuervo N, Grandvaux N. Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology. Viruses. 2016; 8(5):124. https://doi.org/10.3390/v8050124
Chicago/Turabian StyleCervantes-Ortiz, Sandra L., Natalia Zamorano Cuervo, and Nathalie Grandvaux. 2016. "Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology" Viruses 8, no. 5: 124. https://doi.org/10.3390/v8050124
APA StyleCervantes-Ortiz, S. L., Zamorano Cuervo, N., & Grandvaux, N. (2016). Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology. Viruses, 8(5), 124. https://doi.org/10.3390/v8050124