
Hepatitis C Virus Infection Induces Autophagy as a Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response

 , ,
, , 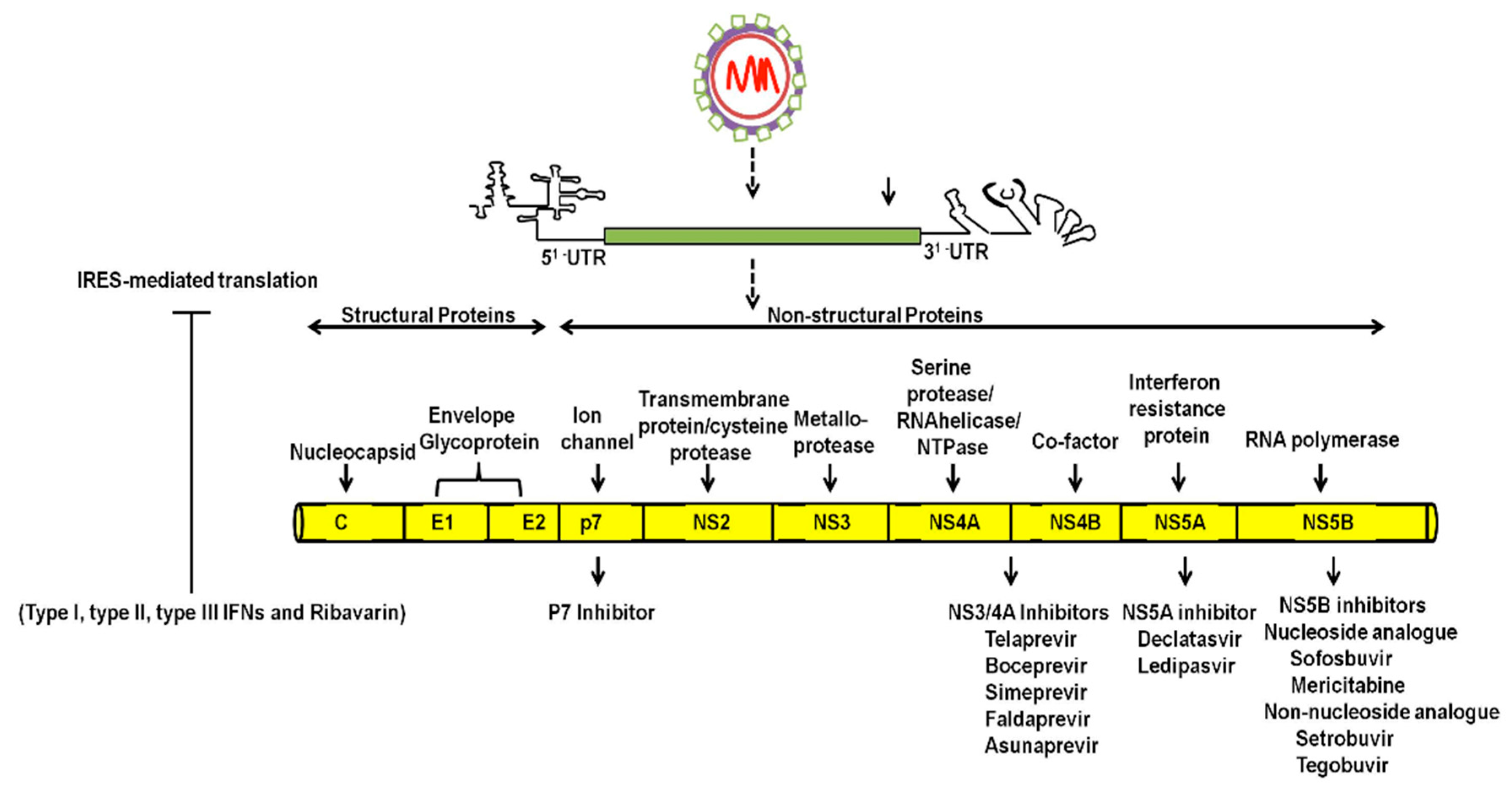{kind=link}
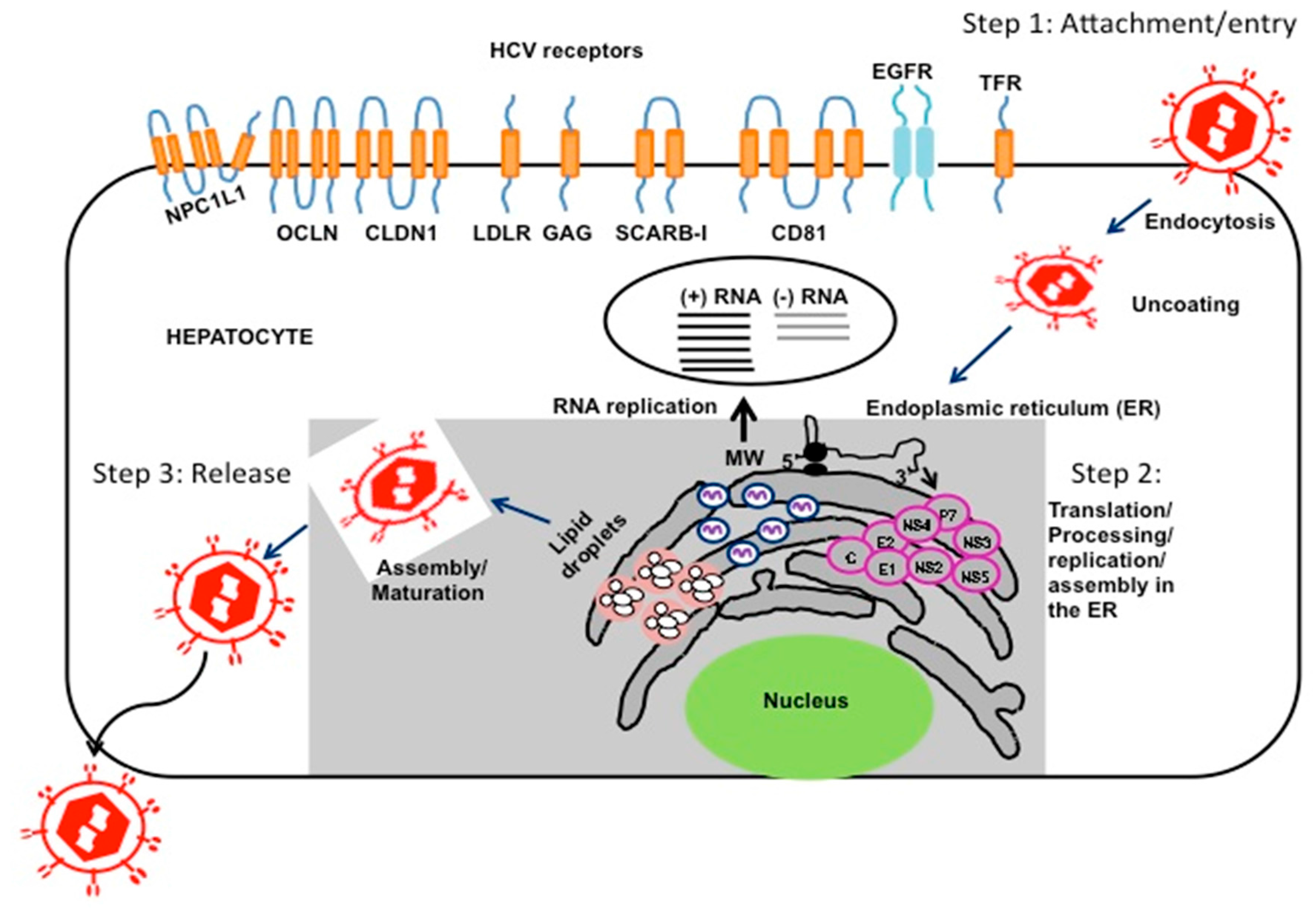{kind=link}
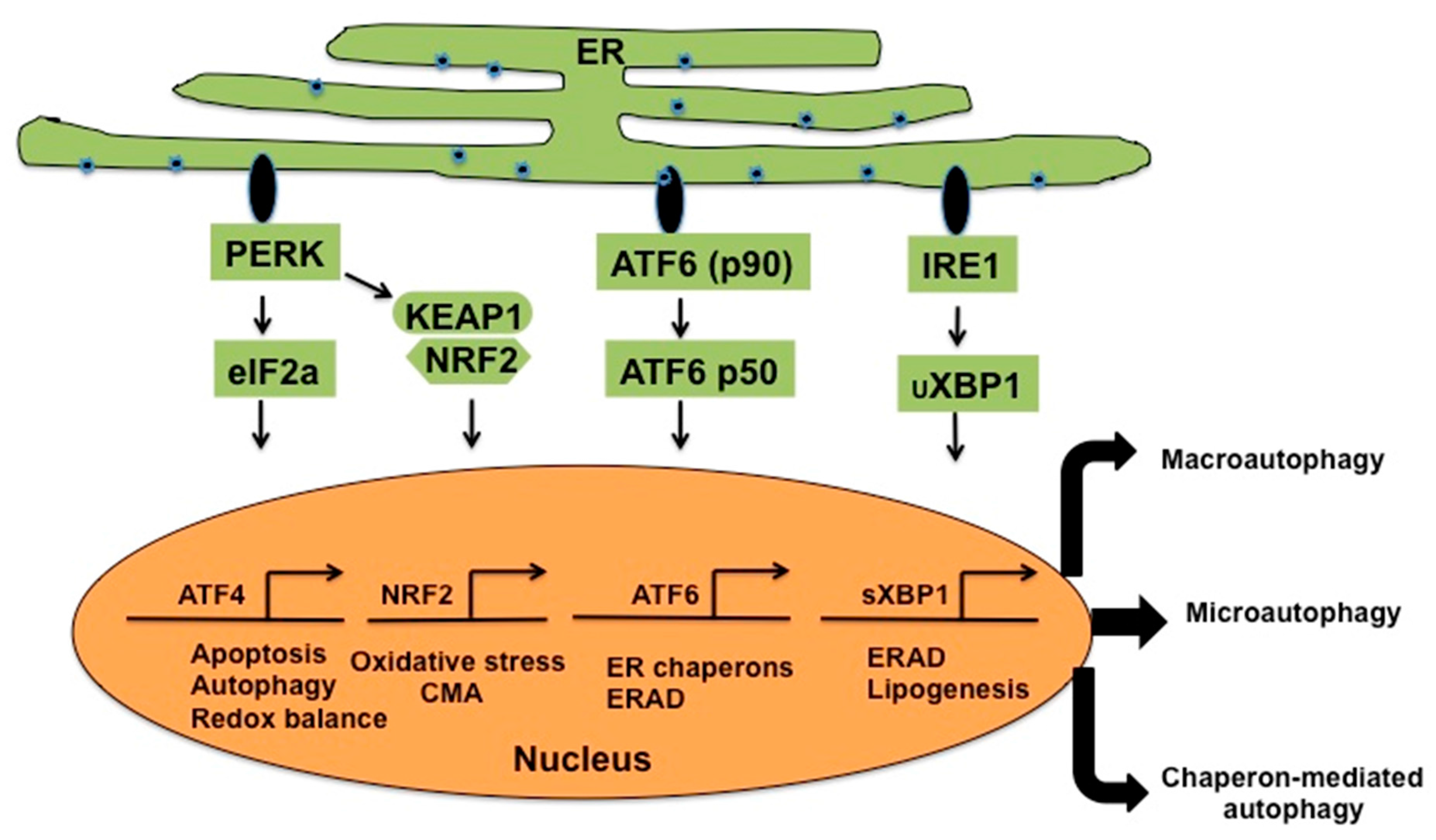{kind=link}
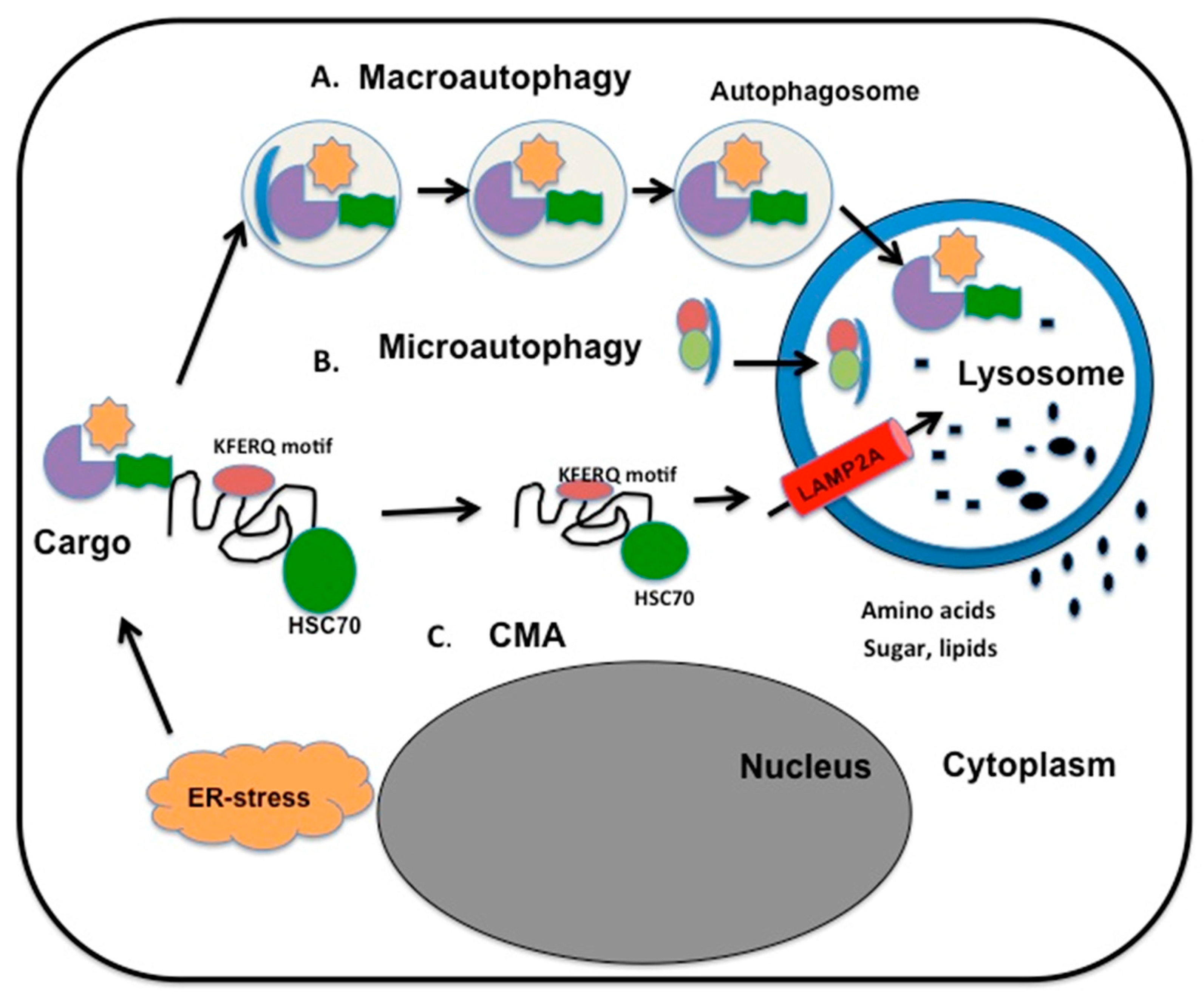{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to HCV Infection and Liver Disease
2. Relationship among Virus Infection, ER-Stress Response and Autophagy
3. Hepatitis C Virus Infection Induces Chronic ER-Stress and UPR
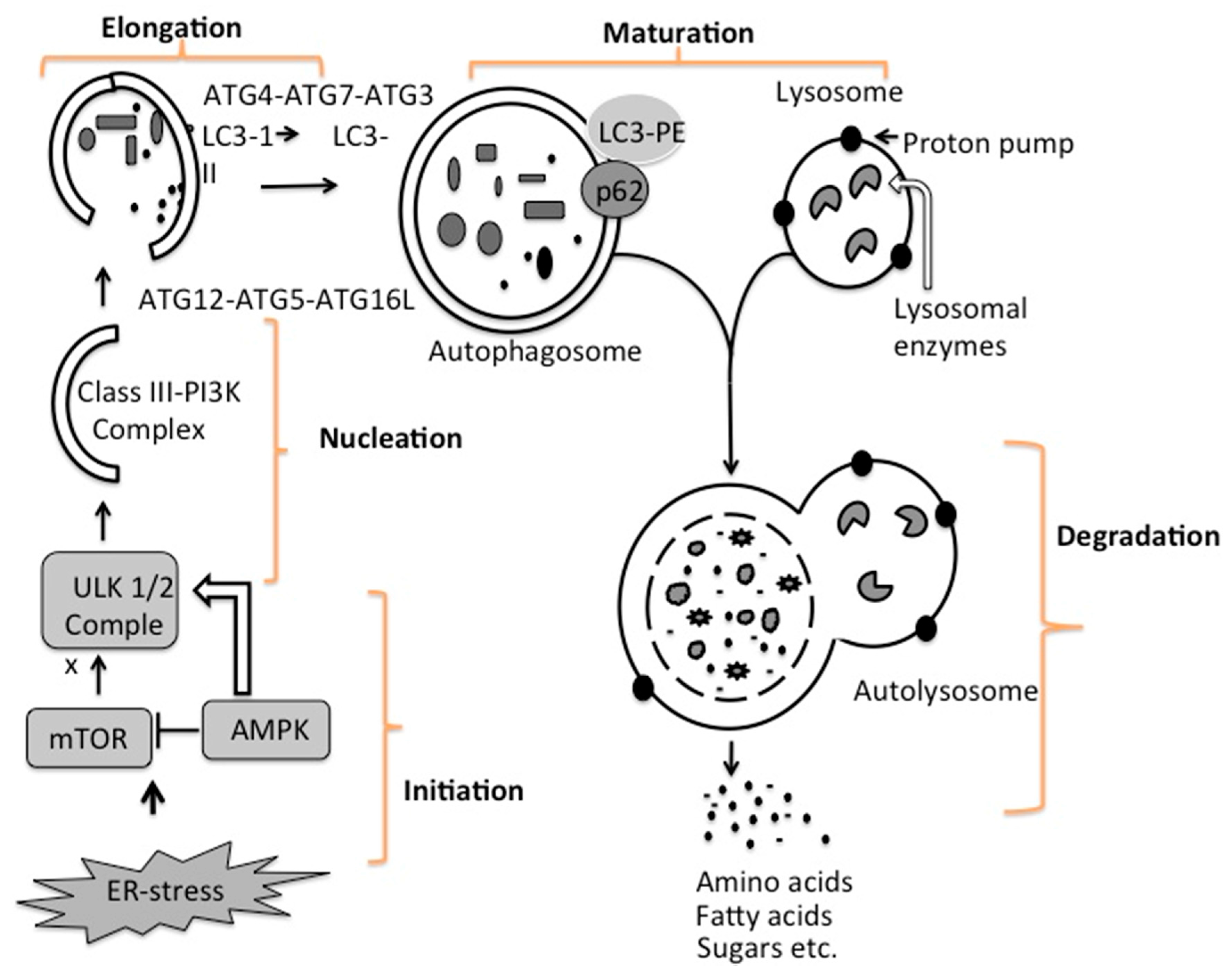
4. Mechanisms of Autophagy Initiation
5. Hepatitis C Virus Infection Promotes Macroautophagy, Mitophagy, and CMA
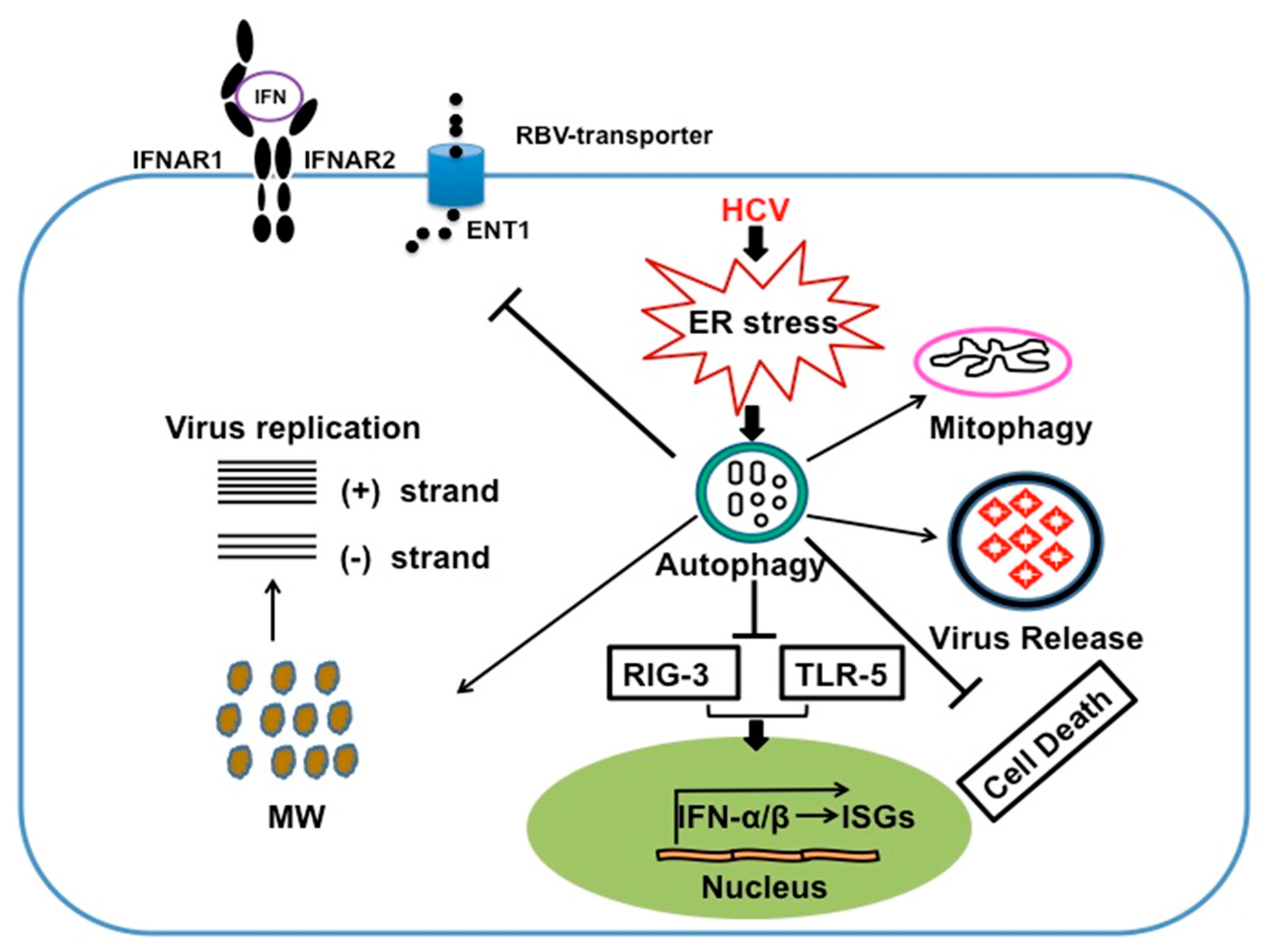
6. Autophagy Serves a Prosurvival Role That Favors Persistent HCV Infection
7. HCV Induces Autophagy Response to Suppress Innate Antiviral Response
8. Autophagy Response Impairs HCV Clearance by IFN-α /RBV Based Antiviral Therapy
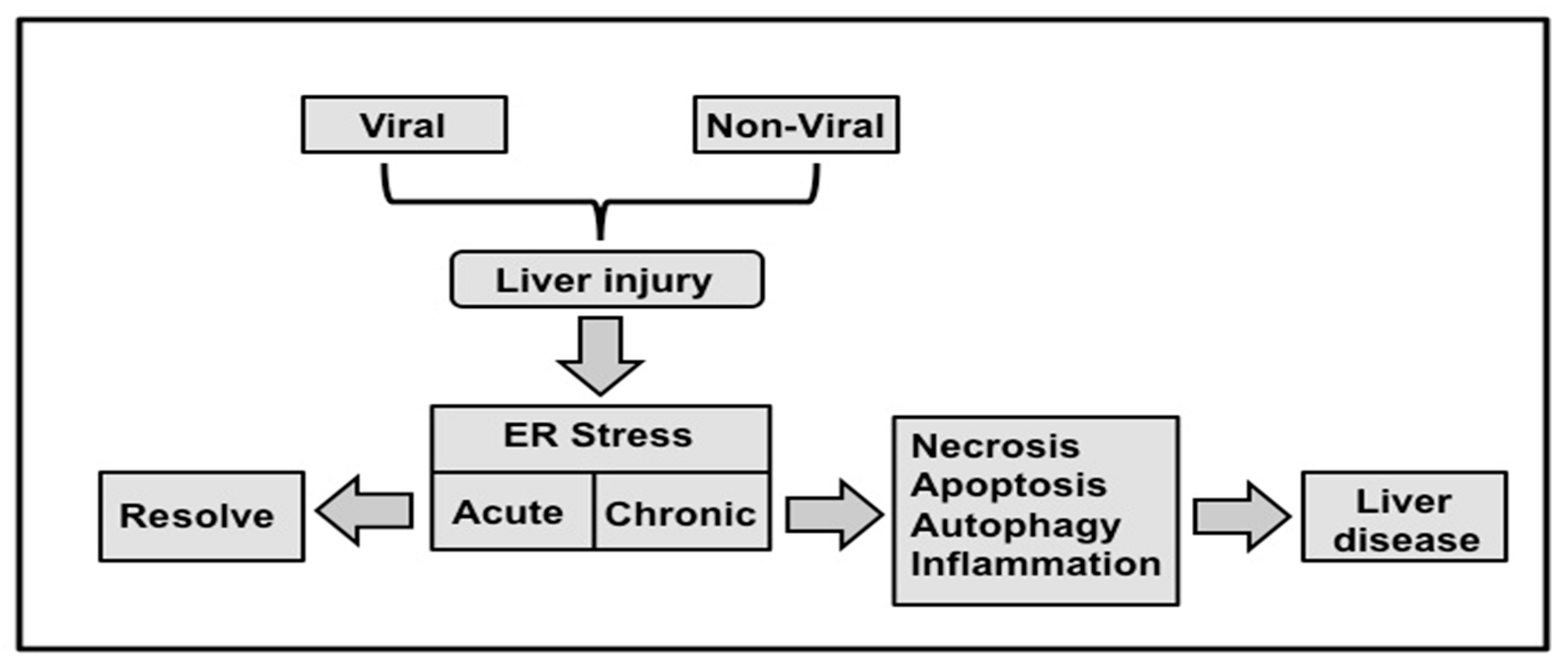
9. Interplay among UPR, Autophagy, Inflammation and Evolution of Chronic Liver Disease
10. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 2005, 5, 558–567. [Google Scholar] [CrossRef]
- Lavanchy, D. The global burden of hepatitis C. Liver Int. Off. J. Int. Assoc. Study Liver 2009, 29, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Kanwal, F.; Davila, J.A.; Kramer, J.; Richardson, P. A new laboratory-based algorithm to predict development of hepatocellular carcinoma in patients with hepatitis C and cirrhosis. Gastroenterology 2014, 146, 1249.e1–1255.e1. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264.e1–1273.e1. [Google Scholar] [CrossRef] [PubMed]
- Buti, M.; Lurie, Y.; Zakharova, N.G.; Blokhina, N.P.; Horban, A.; Teuber, G.; Sarrazin, C.; Balciuniene, L.; Feinman, S.V.; Faruqi, R.; et al. Randomized trial of peginterferon alfa-2b and ribavirin for 48 or 72 weeks in patients with hepatitis C virus genotype 1 and slow virologic response. Hepatology 2010, 52, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M.; Feld, J.J.; Zeuzem, S.; Hoofnagle, J.H. From non-A, non-B hepatitis to hepatitis C virus cure. J. Hepatol. 2015, 62, S87–S99. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.; Pol, S.; Jacobson, I.M.; Marcellin, P.; Gordon, S.C.; Peng, C.Y.; Chang, T.T.; Everson, G.T.; Heo, J.; Gerken, G.; et al. All-oral daclatasvir plus asunaprevir for hepatitis C virus genotype 1b: A multinational, phase 3, multicohort study. Lancet 2014, 384, 1597–1605. [Google Scholar] [CrossRef]
- Lawitz, E.; Sulkowski, M.S.; Ghalib, R.; Rodriguez-Torres, M.; Younossi, Z.M.; Corregidor, A.; DeJesus, E.; Pearlman, B.; Rabinovitz, M.; Gitlin, N.; et al. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: The COSMOS randomised study. Lancet 2014, 384, 1756–1765. [Google Scholar] [CrossRef]
- Mittal, S.; Sada, Y.H.; El-Serag, H.B.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Temporal trends of nonalcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs population. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2015, 13, 594.e1–601.e1. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M. Hepatitis C virus RNA translation. Curr. Top. Microbial. Immunol. 2013, 369, 143–166. [Google Scholar]
- Moradpour, D.; Penin, F. Hepatitis C virus proteins: From structure to function. Curr. Top. Microbial. Immunol. 2013, 369, 113–142. [Google Scholar]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C virus RNA replication and assembly: Living on the fat of the land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, Y.; Seki, N.; Nagano, T.; Abe, H. Chronic hepatitis C virus infection and lipoprotein metabolism. World J. Gastroenterol. 2015, 21, 10299–10313. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Matsuda, M.; Watashi, K.; Aizaki, H.; Matsuura, Y.; Wakita, T.; Suzuki, T. Signal peptidase complex subunit 1 participates in the assembly of hepatitis C virus through an interaction with E2 and NS2. PLoS Pathog. 2013, 9, e1003589. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, J.; Cosset, F.L. Virology and cell biology of the hepatitis C virus life cycle: An update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D. Virion assembly and release. Curr. Top. Microbial. Immunol. 2013, 369, 199–218. [Google Scholar]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; Andre, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Borgese, N. Getting membrane proteins on and off the shuttle bus between the endoplasmic reticulum and the Golgi complex. J. Cell Sci. 2016, 129, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER-stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Helenius, A. Quality controls in the edoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced aoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed]
- He, B. Viruses, ednoplasmic reticulum stress and interferon responses. Cell Death Differ. 2006, 13, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Hedge, N.R.; Chevalier, M.S.; Wisner, T.W.; Denton, M.C.; Shire, K.; Frappier, L.; Johnson, D.C. The role of BiP in endoplasmic reticulum-associated degradation of major histocompatibility complex class I heacy chain induced by cytomegalovirus proteins. J. Biol. Chem. 2006, 281, 20910–20919. [Google Scholar]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, R.L.; Mackenzie, J.M. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune ivasion. J. Virol. 2011, 85, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Pasqual, G.; Burri, D.J.; Pasquato, A.; De La Torre, J.C.; Kunz, S. Role of the host cell’s unfolded protein response in arenavirus infection. J. Virol. 2011, 85, 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Sugden, B. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 2008, 111, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Feng, Z.; He, B. Herpes simplex virus 1 infection activates the endoplamic reticulum resident kinase PERK and mediates eIF-2 alpha dephosphorylation by the gamma (1) 34.5 protein. J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U.; Maini, M.K.; Knolle, P.A. Living in the liver: Hepatic infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Architechure and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Wang, H.; Huang, C.; Huang, Y.; Li, J. Endoplasmic reticulum stress is the crossroads of autophagy, inflammation, and apoptosis signaling pathways and participates in liver fibrosis. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2015, 64, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jheng, J.R.; Ho, J.Y.; Horng, J.T. ER stress, autophagy, and RNA viruses. Front. Microbial. 2014, 5, 388. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Dicks, N.; Gutierrez, K.; Michalak, M.; Bordignon, V.; Agellon, L.B. Endoplasmic reticulum stress, genome damage, and cancer. Front. Oncol. 2015, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Lee, A.S. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J. Cell. Physiol. 2015, 230, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 1999, 339, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Roidl, D.; Hellbach, N.; Bovio, P.P.; Villarreal, A.; Heidrich, S.; Nestel, S.; Gruning, B.A.; Boenisch, U.; Vogel, T. DOT1L Activity Promotes Proliferation and Protects Cortical Neural Stem Cells from Activation of ATF4-DDIT3-Mediated ER Stress in Vitro. Stem Cells 2016, 34, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Vasallo, C.; Gastaminza, P. Cellular stress responses in hepatitis C virus infection: Mastering a two-edged sword. Virus Res. 2015, 209, 100–117. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W. Unfolded protein response in hepatitis C virus infection. Front. Microbial. 2014, 5, 233. [Google Scholar] [CrossRef] [PubMed]
- Tardif, K.D.; Mori, K.; Siddiqui, A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 2002, 76, 7453–7459. [Google Scholar] [CrossRef] [PubMed]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investing. 2011, 121, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, Y.; Imajo, K.; Yoneda, M.; Tomeno, W.; Ogawa, Y.; Kirikoshi, H.; Funakoshi, K.; Ikeda, M.; Kato, N.; Nakajima, A.; et al. Unfolded protein response pathways regulate Hepatitis C virus replication via modulation of autophagy. Biochem. Biophys. Res. Commun. 2013, 432, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Merquiol, E.; Uzi, D.; Mueller, T.; Goldenberg, D.; Nahmias, Y.; Xavier, R.J.; Tirosh, B.; Shibolet, O. HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS ONE 2011, 6, e24660. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.K.; Bao, L.; Song, K.; Aboulnasr, F.M.; Baker, D.P.; Shores, N.; Wimley, W.C.; Liu, S.; Hagedorn, C.H.; Fuchs, S.Y.; et al. HCV infection selectively impairs type I but not type III IFN signaling. Am. J. Pathol. 2014, 184, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Walters, K.A.; Lamb, S.E.; Yeh, M.M.; Zhu, L.F.; Kneteman, N.; Doyle, J.S.; Katze, M.G.; Tyrrell, D.L. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009, 5, e1000291. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Sakamoto, N.; Sekine-Osajima, Y.; Nakagawa, M.; Itsui, Y.; Azuma, S.; Kakinuma, S.; Kiyohashi, K.; Kitazume, A.; Tsuchiya, K.; et al. Cell culture and in vivo analyses of cytopathic hepatitis C virus mutants. Virology 2010, 405, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.K.; Gunduz, F.; Hazari, S.; Kurt, R.; Panigrahi, R.; Poat, B.; Bruce, D.; Cohen, A.J.; Bohorquez, H.E.; Carmody, I.; et al. Impaired expression of type I and type II interferon receptors in HCV-associated chronic liver disease and liver cirrhosis. PLoS ONE 2014, 9, e108616. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Chava, S.; Chandra, P.K.; Balart, L.A.; Lu, K.; Dash, S. Hepatitis C virus replication promotes p53 degradation through chaperon-mediated autophagy. (unpublished; manuscript in preparation).
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [PubMed]
- Asselah, T.; Bieche, I.; Mansouri, A.; Laurendeau, I.; Cazals-Hatem, D.; Feldmann, G.; Bedossa, P.; Paradis, V.; Martinot-Peignoux, M.; Lebrec, D.; et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 2010, 221, 264–274. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; Powell, E.E.; Barrie, H.D.; Clouston, A.D.; McGuckin, M.; Jonsson, J.R. No evidence of the unfolded protein response in patients with chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 2011, 26, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Cheng, J.; Kaplowitz, N. The contribution of ER stress to Liver Disease. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gao, B.; Ye, L.; Han, X.; Wang, W.; Kong, L.; Fang, X.; Zeng, Y.; Zheng, H.; Li, S.; et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP pathway of unfolded protein response. Virus Res. 2007, 124, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Christen, V.; Treves, S.; Duong, F.H.; Heim, M.H. Activation of endoplasmic reticulum stress response by hepatitis C virus up-regulates protein phosphatase 2A. Hepatology 2007, 46, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N. Hyperhomocysteminemia, endoplasmic reticulum stress and alcoholic liver injury. World J. Gastroenterol. 2004, 10, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E. Regulation of hepatic lipogenesis by transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Choe, S.S.; Shin, K.C. Endoplasmic reticulum stress induces hepatis steatosis via increased expression of hepatic low-density lipoprotein receptor. Hepatology 2014, 57, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Bailly-Maitre, B.; Fondevila, C.; Kaldas, F.; Droin, N.; Luciano, F.; Ricci, J.E.; Croxton, R.; Krajewska, M.; Zapata, J.M.; Kupiec-Weqlinski, J.W.; et al. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic stress and ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2006, 103, 2809–2814. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.; Payne, C.M.; Bernstein, C.; Schneider, J.; Beard, S.E.; Crowley, C.L. Activation of the promoters of genes associated with DNA damage, oxidative stress, ER-stress and protein malfolding by the bile salt, deoxycholate. Toxicol. Lett. 1999, 108, 37–46. [Google Scholar] [CrossRef]
- Ren, F.; Shi, H.; Zhang, L.; Zhang, X.; Wen, T.; Xie, B.; Zheng, S.; Li, L.; Chen, D.; Duan, Z. The desregulation of endoplasmic reticulum stress response in acute-on-chronic liver failure patients caused by acute excerbation of chronic hepatitis B. J. Viral Hepat. 2016, 23, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross talk between ER-stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 2015, 1292, 205–214. [Google Scholar] [PubMed]
- Vescovo, T.; Romagnoli, A.; Perdomo, A.B.; Corazzari, M.; Ciccosanti, F.; Alonzi, T.; Nardacci, R.; Ippolito, G.; Tripodi, M.; Garcia-Monzon, C.; et al. Autophagy protects cells from HCV induced defect in lipid metabolism. Gastroenterology 2012, 142, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Kardon, T.; Wunderlich, L.; Szarka, A.; Kiss, A.; Schaff, Z.; Banhegyi, G.; Mandl, J. Acetaminophen induces ER-dependnet signaling in mouse liver. Arch Biochem. Biophys. 2007, 459, 273–279. [Google Scholar] [PubMed]
- Lake, A.D.; Novak, P.; Hardwick, R.N.; Flores-Keown, B.; Zhao, F.; Klimecki, W.T.; Cherrington, N.J. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol. Sci. Off. J. Soc. Toxicol. 2014, 137, 26–35. [Google Scholar]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, D.; Crespo, I.; San-Miguel, B.; Alvarez, M.; Prieto, J.; Tunon, M.J.; Gonzalez-Gallego, J. Autophagic response in the Rabbit Hemorrhagic Disease, an animal model of virally-induced fulminant hepatic failure. Vet. Res. 2014, 45, 15. [Google Scholar] [PubMed]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [PubMed]
- Crawford, S.E.; Hyser, J.M.; Utama, B.; Estes, M.K. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-beta signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. USA 2012, 109, E3405–E3413. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Werneke, S.W.; de la Calle, C.; Guivel-Benhassine, F.; Giodini, A.; Peduto, L.; Levine, B.; Schwartz, O.; Lenschow, D.J.; Albert, M.L. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 2012, 209, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [PubMed]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Ramiere, C.; Rodriguez, J.; Enache, L.S.; Lotteau, V.; Andre, P.; Diaz, O. Activity of hexokinase is increased by its interaction with hepatitis C virus protein NS5A. J. Virol. 2014, 88, 3246–3254. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Shoji, I.; Ogawa, W.; Kaneda, S.; Soga, T.; Jiang, D.P.; Ide, Y.H.; Hotta, H. Hepatitis C virus infection promotes hepatic gluconeogenesis through an NS5A-mediated, FoxO1-dependent pathway. J. Virol. 2011, 85, 8556–8568. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed]
- Kurt, R.; Chandra, P.K.; Aboulnasr, F.; Panigrahi, R.; Ferraris, P.; Aydin, Y.; Reiss, K.; Wu, T.; Balart, L.A.; Dash, S. Chaperone-Mediated Autophagy Targets IFNAR1 for Lysosomal Degradation in Free Fatty Acid Treated HCV Cell Culture. PLoS ONE 2015, 10, e0125962. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.W.; Peng, Z.J.; Ren, G.F.; Wang, G.X. The different roles of selective autophagic protein degradation in mammalian cells. Oncotarget 2015, 6, 37098–37116. [Google Scholar] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Cuervo, A.M. Chaperone-mediated autophagy in protein quality control. Curr. Opin. Cell Biol. 2011, 23, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Chava, S.; Chandra, P.K.; Aydin, Y.; Balart, L.A.; Wu, T. Autophagy in hepatocellular carcinomas: From pathophysiology to therapeutic response. Hepatic Med. Evid. Res. 2016, 8, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Fenouille, N.; Auberger, P. When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res. 2012, 2, 397–413. [Google Scholar] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structure for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Chiramel, A.I.; Brandy, N.R.; Bartenschalager, R. Divergent role of autophagy in virus infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Lazar, C.; Uta, M.; Branza-Nichita, N. Modulation of the unfolded protein response by human hepatitis B virus. Front. Microbiol. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shelly, S.; Lukinova, N.; Bambina, S.; Berman, A.; Cherry, S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009, 30, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.E.; Jackson, W.; Benetti, L.; Grose, C. Autophagosome formation during varicella-zoster virus infection following endoplasmic stress and the unfolded protein response. J. Virol. 2011, 85, 9414–9424. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tian, Y.; Ou, J.H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Kang, R.; Wang, J.; Luo, G.; Yang, W.; Zhao, Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013, 9, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, R.; Chandra, P.K.; Ferraris, P.; Kurt, R.; Song, K.; Garry, R.F.; Reiss, K.; Coe, I.R.; Furihata, T.; Balart, L.A.; et al. Persistent hepatitis C virus infection impairs ribavirin antiviral activity through clathrin-mediated trafficking of equilibrative nucleoside transporter 1. J. Virol. 2015, 89, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011, 85, 13185–13194. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.C.; Bhattacharya, S.; Nomoto, A.; Lin, J.; Zaidi, S.K.; Oberley, T.D.; Weinman, S.A.; Azhar, S.; Huang, T.T. Persistent expression of hepatitis C virus non-structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS ONE 2011, 6, e28551. [Google Scholar] [CrossRef] [PubMed]
- Rautou, P.E.; Cazals-Hatem, D.; Feldmann, G.; Mansouri, A.; Grodet, A.; Barge, S.; Martinot-Peignoux, M.; Duces, A.; Bieche, I.; Lebrec, D.; et al. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am. J. Pathol. 2011, 178, 2708–2715. [Google Scholar] [CrossRef] [PubMed]
- Mohl, B.P.; Tedbury, P.R.; Griffin, S.; Harris, M. Hepatitis C virus-induced autophagy is independent of the unfolded protein response. J. Virol. 2012, 86, 10724–10732. [Google Scholar] [CrossRef] [PubMed]
- Su, W.C.; Chao, T.C.; Huang, Y.L.; Weng, S.C.; Jeng, K.S.; Lai, M.M. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef] [PubMed]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Tsurumi, T. Genome guardian p53 and viral infections. Rev. Med. Virol. 2013, 23, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Zaika, A.I.; Wei, J.; Noto, J.M.; Peek, R.M. Microbial Regulation of p53 Tumor Suppressor. PLoS Pathog. 2015, 11, e1005099. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.C. Structure and function of the p53 tumor suppressor gene: Clues for rational cancer therapeutic strategies. J. Natl. Cancer Inst. 1996, 88, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.H.; Clarke, M.F. Regulation of p53 localization. Eur. J. Biochem./FEBS 2001, 268, 2779–2783. [Google Scholar] [CrossRef]
- Lu, W.; Lo, S.Y.; Chen, M.; Wu, K.; Fung, Y.K.; Ou, J.H. Activation of p53 tumor suppressor by hepatitis C virus core protein. Virology 1999, 264, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kato, N.; Lan, K.; Yoshida, H.; Kato, J.; Goto, T.; Shiratori, Y.; Omata, M. Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J. Biol. Chem. 2000, 275, 34122–34130. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.B.; Steele, R.; Meyer, K.; Ray, R. Hepatitis C virus core protein represses p21WAF1/Cip1/Sid1 promoter activity. Gene 1998, 208, 331–336. [Google Scholar] [CrossRef]
- Kao, C.F.; Chen, S.Y.; Chen, J.Y.; Wu Lee, Y.H. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene 2004, 23, 2472–2483. [Google Scholar] [CrossRef] [PubMed]
- Ishido, S.; Hotta, H. Complex formation of the nonstructural protein 3 of hepatitis C virus with the p53 tumor suppressor. FEBS Lett. 1998, 438, 258–262. [Google Scholar] [CrossRef]
- Kwun, H.J.; Jung, E.Y.; Ahn, J.Y.; Lee, M.N.; Jang, K.L. p53-dependent transcriptional repression of p21(waf1) by hepatitis C virus NS3. J. Gen. Virol. 2001, 82, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Nagano-Fujii, M.; Tanaka, M.; Nomura-Takigawa, Y.; Ikeda, M.; Kato, N.; Sada, K.; Hotta, H. NS3 protein of Hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J. Gen. Virol. 2006, 87, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Nagano-Fujii, M.; Deng, L.; Ishido, S.; Sada, K.; Hotta, H. Single-point mutations of hepatitis C virus NS3 that impair p53 interaction and anti-apoptotic activity of NS3. Biochem. Biophys. Res. Commun. 2006, 340, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 2001, 75, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.H.; Sheu, M.L.; Hwang, S.J.; Yen, S.H.; Chen, S.Y.; Wu, J.C.; Wang, Y.J.; Kato, N.; Omata, M.; Chang, F.Y.; et al. HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene 2002, 21, 4801–4811. [Google Scholar] [CrossRef] [PubMed]
- Qadri, I.; Iwahashi, M.; Simon, F. Hepatitis C virus NS5A protein binds TBP and p53, inhibiting their DNA binding and p53 interactions with TBP and ERCC3. Biochim. Biophys. Acta 2002, 1592, 193–204. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Steele, R.; Meyer, K.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A protein modulates cell cycle regulatory genes and promotes cell growth. J. Gen. Virol. 1999, 80, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Bressac, B.; Galvin, K.M.; Liang, T.J.; Isselbacher, K.J.; Wands, J.R.; Ozturk, M. Abnormal structure and expression of p53 gene in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 1990, 87, 1973–1977. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Levine, B. Eating the enemy within: Autophagy in infectious diseases. Cell Death Differ. 2009, 16, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randal, G. Dengue virus and autophagy. Viruses 2011, 3, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. Antigen processing by macroautophagy for MHC presentation. Front. Immunol. 2011, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Munz, C. MHC presentation via autophagy and how viruses escape from it. Semin. Immunopathol. 2010, 32, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, M.A.; Deretic, V. Toll-like receptors in control of immunological autophagy. Cell Death Differ. 2009, 16, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Into, T.; Inomata, M.; Takayama, E.; Takigawa, T. Autophagy in regulation of Toll-like receptor signaling. Cell. Signal. 2012, 24, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Katze, M.G.; Fornek, J.L.; Palermo, R.E.; Walters, K.A.; Korth, M.J. Innate immune modulation by RNA viruses: Emerging insights from functional genomics. Nat. Rev. Immunol. 2008, 8, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Navajas, J.M.; Lee, J.; David, M.; Raz, E. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 2012, 12, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.-T.; Chen, S.S.-L. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell. Mol. Immunol. 2016, 13, 11–35. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A. A new paradigm: Innate immune sensing of viruses via the unfolded protein response. Front. Microbiol. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jilg, N.; Shao, R.X.; Lin, W.; Fusco, D.N.; Zhao, H.; Goto, K.; Peng, L.F.; Chen, W.C.; Chung, R.T. (2011) IL28B inhibits hepatitis C virus replication through the Jak-Stat pathway. J. Hepatol. 2011, 55, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Guo, H.; Xu, C.; Chang, J.; Gu, B.; Wang, L.; Block, T.M.; Guo, J.T. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 2008, 82, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Prabhu, R.; Hazari, S.; Bastian, F.; Garry, R.F.; Zou, W.; Haque, S.; Joshi, V.; Regenstein, F.G.; Thung, S.N. Interferon alpha, beta, gamma each inhibits hepatitis C virus replication at the levels of internal ribosome entry site mediated translation. Liver Int. 2005, 25, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Hazari, S.; Patel, A.; Prabhu, R.; Bastian, F.; Garry, R.; Joshi, V.; Haque, S.; Regenstein, F.G.; Elliot, R.; Dash, S. Interferon alpha inhibits internal ribosome entry site mediated translation of green fluorescence protein from six different HCV genotypes. J. Gen. Virol. 2005, 86, 3047–3053. [Google Scholar] [CrossRef] [PubMed]
- Poat, B.; Hazari, S.; Chandra, P.K.; Gunduz, F.; Garry, R.F.; Dash, S. Intracellular expression of IRF-9 Stat fusion protein overcome defective Jak-Stat signaling and inhibits HCV RNA replication. Virol. J. 2010, 7, 265. [Google Scholar] [CrossRef] [PubMed]
- Poat, B.; Hazari, S.; Chandra, P.; Gunduz, F.; Balart, L.; Dash, S. SH2 modified STAT1 induces HLA-1 expression and improves IFN-signaling and inhibits HCV RNA replication. PLoS ONE 2010, 5, e13117. [Google Scholar] [CrossRef]
- Panigrahi, R.; Hazari, S.; Chandra, S.; Chandra, P.K.; Datta, S.; Kurt, R.; Cameron, C.E.; Huang, Z.; Zhang, H.; Garry, R.F.; et al. Interferon and ribavirin combination treatment synergistically inhibit HCV internal ribosome entry site mediated translation at the level of polyribosome formation. PLoS ONE 2013, 8, e72791. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Hazari, S.; Chandra, P.K.; Samara, M.; Poat, B.; Gunduz, F.; Wimley, W.C.; Hauser, H.; Koster, M.; Lamaze, C.; et al. Mechanisms of HCV’s resistance to IFN-α in cell culture involves expression of functional IFN-α receptor 1. Virol. J. 2011, 8, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huangfu, W.C.; Kumar, K.G.; Qian, J.; Casey, J.P.; Hamanaka, R.B.; Grigoriadou, C.; Aldabe, R.; Diehl, J.A.; Fuchs, S.Y. Virus induced unfolded protein response attenuates antiviral defense via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 2009, 5, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Dice, J.F. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J. Biol. Chem. 1988, 263, 6797–6805. [Google Scholar] [PubMed]
- Dice, J. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. 1990, 15, 305–309. [Google Scholar] [CrossRef]
- Cuervo, A.; Dice, J. A receptor for selective uptake and degradation of proteins by lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte death: A clear and present danger. Physiol. Rev. 2010, 90, 1165–1194. [Google Scholar] [CrossRef] [PubMed]
- Canbay, A.; Friedman, S.; Gores, G.J. Apoptosis: The nexus of liver injury and fibrosis. Hepatology 2004, 39, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. That which does not kill me makes me stronger: Adapting to chronic ER stress. Trends Biochem. Sci. 2007, 32, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, S.; Mandrekar, P. Cellular stress response and innate immune signaling; integrating pathways in host defense and inflammation. J. Leukoc. Biol. 2013, 94, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Zenetti, M.; Rodvold, J.J.; Mahadevan, N.R. The evolving paragigm of cell-nonautonomous UPR-based regulation of immunity by cancer cells. Oncogene 2016, 35, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Rusyn, I.; Lemon, S.M. Mechanisms of HCV-induced liver cancer: What did we learn from in vitro and animal studies? Cancer Lett. 2014, 345, 210–215. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Lemon, S.M. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 2011, 30, 1969–1983. [Google Scholar] [CrossRef] [PubMed]
- Sipeki, N.; Antal-Szalmas, P.; Lakatos, P.L.; Papp, M. Immune dysfunction in cirrhosis. World J. Gastroenterol. 2014, 20, 2564–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravalli, R.N. Role of innate immunity in the development of hepatocellular carcinoma. World J. Gastroenterol. 2013, 19, 7500–7514. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, J.K.; Scarzello, A.J.; Jiang, Q.; Wiltrout, R.H. Chronic inflammation, immune escape, and oncogenesis in the liver: A unique neighborhood for novel intersections. Hepatology 2012, 56, 1567–1574. [Google Scholar] [CrossRef] [PubMed]








© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, S.; Chava, S.; Aydin, Y.; Chandra, P.K.; Ferraris, P.; Chen, W.; Balart, L.A.; Wu, T.; Garry, R.F. Hepatitis C Virus Infection Induces Autophagy as a Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response. Viruses 2016, 8, 150. https://doi.org/10.3390/v8050150
Dash S, Chava S, Aydin Y, Chandra PK, Ferraris P, Chen W, Balart LA, Wu T, Garry RF. Hepatitis C Virus Infection Induces Autophagy as a Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response. Viruses. 2016; 8(5):150. https://doi.org/10.3390/v8050150
Chicago/Turabian StyleDash, Srikanta, Srinivas Chava, Yucel Aydin, Partha K. Chandra, Pauline Ferraris, Weina Chen, Luis A. Balart, Tong Wu, and Robert F. Garry. 2016. "Hepatitis C Virus Infection Induces Autophagy as a Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response" Viruses 8, no. 5: 150. https://doi.org/10.3390/v8050150
APA StyleDash, S., Chava, S., Aydin, Y., Chandra, P. K., Ferraris, P., Chen, W., Balart, L. A., Wu, T., & Garry, R. F. (2016). Hepatitis C Virus Infection Induces Autophagy as a Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response. Viruses, 8(5), 150. https://doi.org/10.3390/v8050150