
Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway

Abstract
:1. Introduction
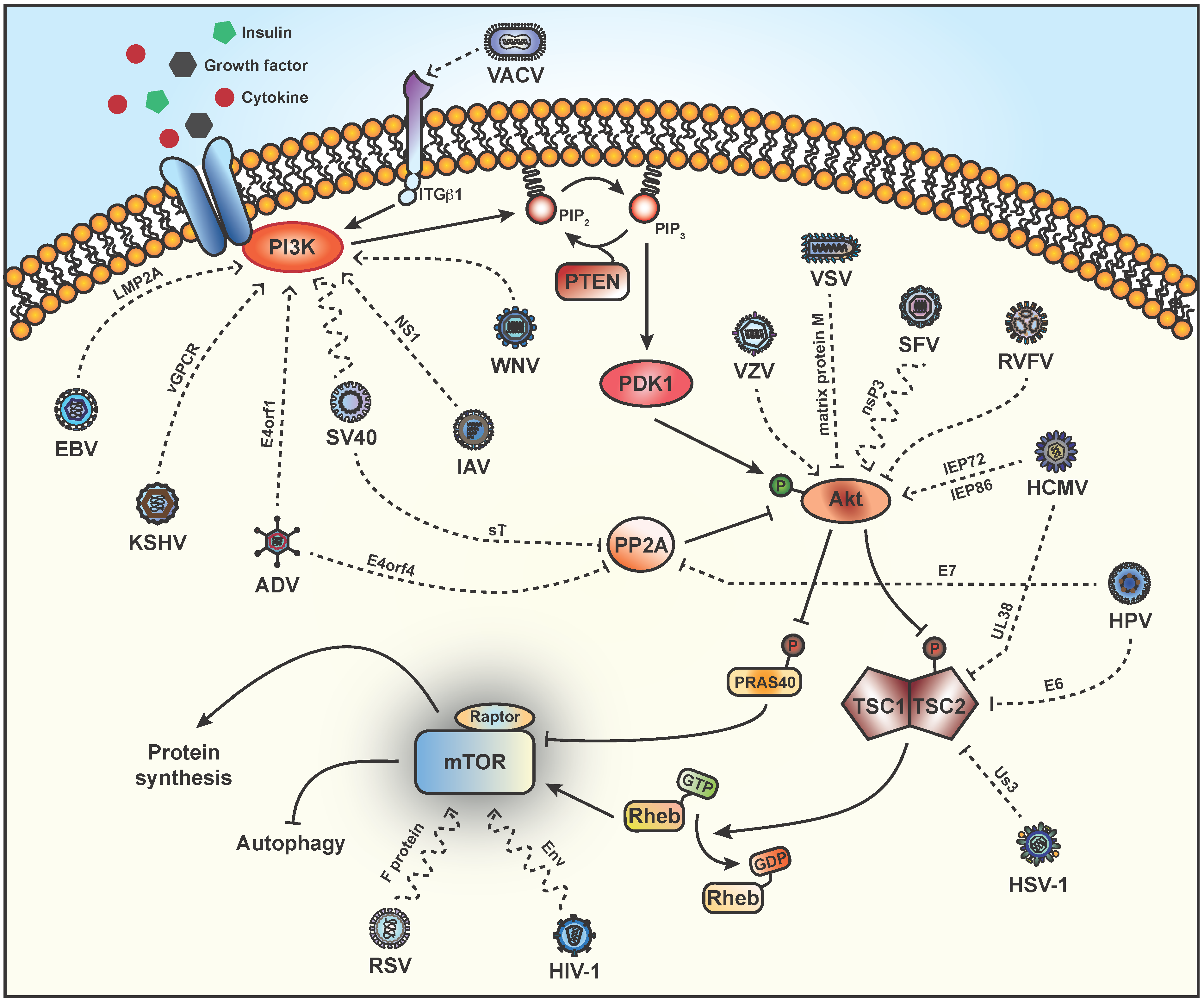
2. Stimulation of PI3K
3. Activation of Akt
4. TSC2
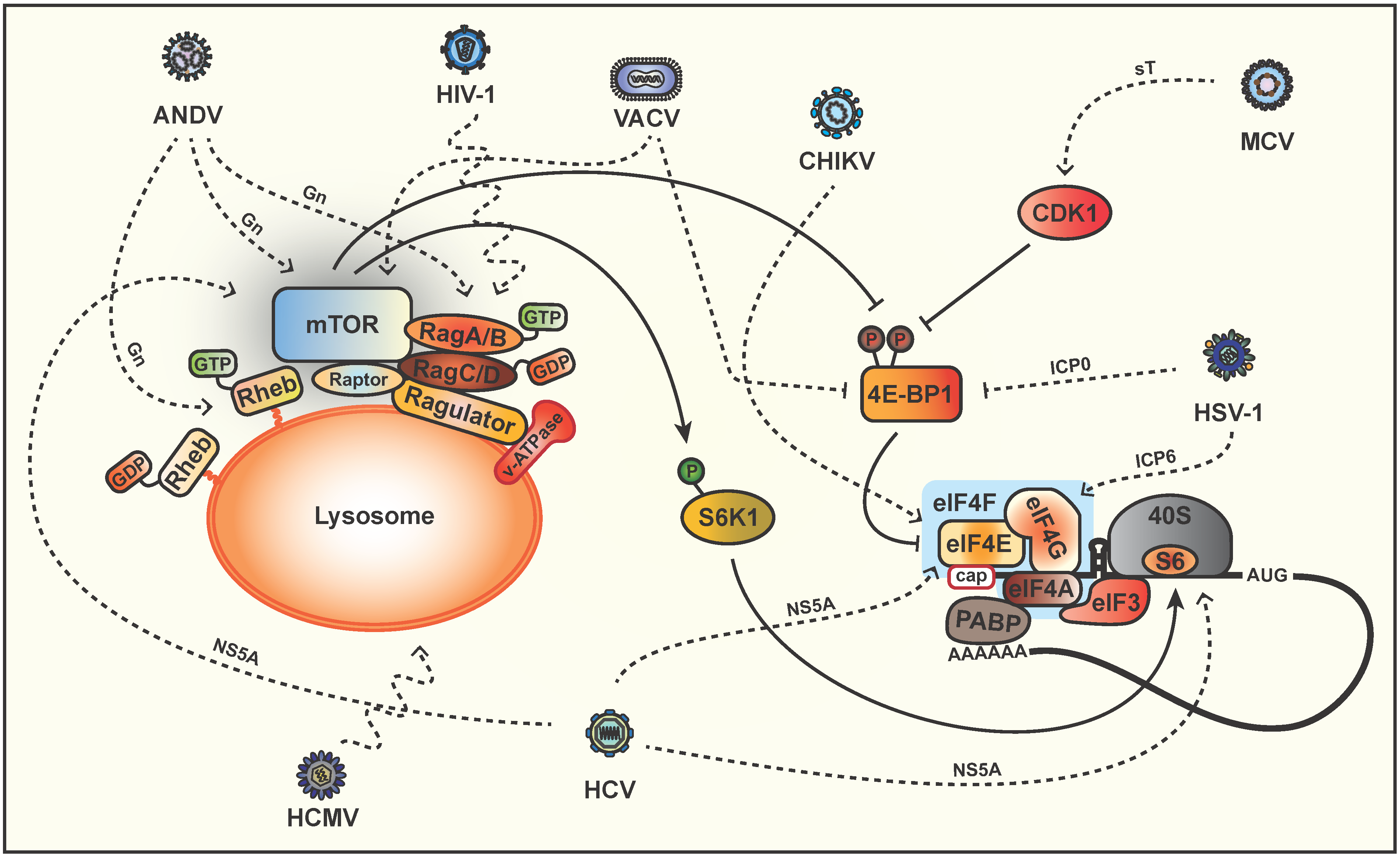
5. Phosphorylation of Downstream Targets
6. Modulation of mTOR via Lysosomal Signaling
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Foster, K.G.; Fingar, D.C. Mammalian target of rapamycin (mTOR): Conducting the cellular signaling symphony. J. Biol. Chem. 2010, 285, 14071–14077. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. The pharmacology of mTOR inhibition. Sci. Signal. 2009, 2, pe24. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Meyers, R.E.; Cantley, L.C. Phosphoinositide kinases. Annu. Rev. Biochem. 1998, 67, 481–507. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Sabatini, D.M. Nutrients and growth factors in mTORC1 activation. Biochem. Soc. Trans. 2013, 41, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Proud, C.G. mTORC1 signaling: What we still don’t know. J. Mol. Cell Biol. 2011, 3, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.K.; Latorre, I.J.; Chung, S.H.; Caruana, G.; Bernstein, A.; Jones, S.N.; Donehower, L.A.; Justice, M.J.; Garner, C.C.; Javier, R.T. Oncogenic function for the Dlg1 mammalian homolog of the Drosophila discs-large tumor suppressor. EMBO J. 2006, 25, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.; Kumar, M.; Taruishi, M.; Javier, R.T. The human adenovirus E4-ORF1 protein subverts discs large 1 to mediate membrane recruitment and dysregulation of phosphatidylinositol 3-kinase. PLoS Pathog. 2014, 10, e1004102. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kong, K.; Javier, R.T. Hijacking Dlg1 for oncogenic phosphatidylinositol 3-kinase activation in human epithelial cells is a conserved mechanism of human adenovirus E4-ORF1 proteins. J. Virol. 2014, 88, 14268–14277. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, C.; Klupsch, K.; Choi, S.; Bagus, B.; Soria, C.; Shen, J.; McCormick, F.; Stokoe, D. Adenoviral proteins mimic nutrient/growth signals to activate the mTOR pathway for viral replication. EMBO J. 2005, 24, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Scott, R.S.; Amirghahari, N.; Nathan, C.O.; Young, L.S.; Dawson, C.W.; Sixbey, J.W. Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J. Virol. 2005, 79, 5499–5506. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Alwine, J.C. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3'-OH kinase pathway and the cellular kinase Akt. J. Virol. 2002, 76, 3731–3738. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Perez, C.; Notary, J.; Mohr, I. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J. Virol. 2005, 79, 8057–8064. [Google Scholar] [CrossRef] [PubMed]
- Moorman, N.J.; Cristea, I.M.; Terhune, S.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 2008, 3, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Alwine, J.C. Dynein mediates the localization and activation of mTOR in normal and human cytomegalovirus-infected cells. Genes Dev. 2012, 26, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Hu, X.; Li, Y.; Zheng, L.; Zhou, Y.; Jiang, H.; Ning, T.; Basang, Z.; Zhang, C.; Ke, Y. Human papillomavirus 16 E6 oncoprotein interferences with insulin signaling pathway by binding to tuberin. J. Biol. Chem. 2004, 279, 35664–35670. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Ding, H.; Lu, Z.; Li, Y.; Pan, Y.; Ning, T.; Ke, Y. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells 2008, 13, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef] [PubMed]
- Chuluunbaatar, U.; Roller, R.; Feldman, M.E.; Brown, S.; Shokat, K.M.; Mohr, I. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 2010, 24, 2627–2639. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mohr, I. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 2004, 18, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mohr, I. Assembly of an active translation initiation factor complex by a viral protein. Genes Dev. 2006, 20, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Montaner, S.; Sodhi, A.; Pece, S.; Mesri, E.A.; Gutkind, J.S. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 2001, 61, 2641–2648. [Google Scholar] [PubMed]
- Sodhi, A.; Chaisuparat, R.; Hu, J.; Ramsdell, A.K.; Manning, B.D.; Sausville, E.A.; Sawai, E.T.; Molinolo, A.; Gutkind, J.S.; Montaner, S. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell 2006, 10, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.; Jurczak, A.; Cheng, L.A.; Baker, D.C.; Benjamin, T.L. Evidence of a role for phosphatidylinositol 3-kinase activation in the blocking of apoptosis by polyomavirus middle T antigen. J. Virol. 1998, 72, 3221–3226. [Google Scholar] [PubMed]
- Utermark, T.; Schaffhausen, B.S.; Roberts, T.M.; Zhao, J.J. The p110alpha isoform of phosphatidylinositol 3-kinase is essential for polyomavirus middle T antigen-mediated transformation. J. Virol. 2007, 81, 7069–7076. [Google Scholar] [CrossRef] [PubMed]
- Werden, S.J.; Barrett, J.W.; Wang, G.; Stanford, M.M.; McFadden, G. M-T5, the ankyrin repeat, host range protein of myxoma virus, activates Akt and can be functionally replaced by cellular PIKE-A. J. Virol. 2007, 81, 2340–2348. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Barrett, J.W.; Stanford, M.; Werden, S.J.; Johnston, J.B.; Gao, X.; Sun, M.; Cheng, J.Q.; McFadden, G. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc. Natl. Acad. Sci. USA 2006, 103, 4640–4645. [Google Scholar] [CrossRef] [PubMed]
- Pallas, D.C.; Shahrik, L.K.; Martin, B.L.; Jaspers, S.; Miller, T.B.; Brautigan, D.L.; Roberts, T.M. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 1990, 60, 167–176. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Collins, C.; Fried, M. Polyoma and SV40 proteins differentially regulate PP2A to activate distinct cellular signaling pathways involved in growth control. Proc. Natl. Acad. Sci. USA 2006, 103, 19290–19295. [Google Scholar] [CrossRef] [PubMed]
- Izmailyan, R.; Hsao, J.C.; Chung, C.S.; Chen, C.H.; Hsu, P.W.; Liao, C.L.; Chang, W. Integrin beta1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 2012, 86, 6677–6687. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Arias, C.; Perez, C.; Halladin, D.; Escandon, M.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Mohr, I. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol. Cell. Biol. 2008, 28, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Rahaus, M.; Desloges, N.; Wolff, M.H. Varicella-zoster virus requires a functional PI3K/Akt/GSK-3alpha/beta signaling cascade for efficient replication. Cell. Signal. 2007, 19, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhu, S.; Wang, J.; Liu, J. Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway during porcine circovirus type 2 infection facilitates cell survival and viral replication. J. Virol. 2012, 86, 13589–13597. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.R.; Chiu, H.C.; Liao, T.L.; Chuang, K.P.; Shih, W.L.; Liu, H.J. Avian Reovirus Protein p17 Functions as a Nucleoporin Tpr Suppressor Leading to Activation of p53, p21 and PTEN and Inactivation of PI3K/AKT/mTOR and ERK Signaling Pathways. PLoS ONE 2015, 10, e0133699. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhang, Y.; Jia, L.; Wu, H.; Fan, C.; Sun, Y.; Ye, C.; Liao, M.; Zhou, J. Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy 2015, 11, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Basantray, I.; Mamidi, P.; Nayak, T.K.; Pratheek, B.M.; Chattopadhyay, S.; Chattopadhyay, S. Heat shock protein 90 positively regulates Chikungunya virus replication by stabilizing viral non-structural protein nsP2 during infection. PLoS ONE 2014, 9, e100531. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Stapleford, K.; Guivel-Benhassine, F.; Vignuzzi, M.; Schwartz, O.; Albert, M.L. Inhibition of mTORC1 Enhances the Translation of Chikungunya Proteins via the Activation of the MnK/eIF4E Pathway. PLoS Pathog. 2015, 11, e1005091. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Werneke, S.W.; de la Calle, C.; Guivel-Benhassine, F.; Giodini, A.; Peduto, L.; Levine, B.; Schwartz, O.; Lenschow, D.J.; Albert, M.L. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 2012, 209, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Thaa, B.; Biasiotto, R.; Eng, K.; Neuvonen, M.; Gotte, B.; Rheinemann, L.; Mutso, M.; Utt, A.; Varghese, F.; Balistreri, G.; et al. Differential Phosphatidylinositol-3-Kinase-Akt-mTOR Activation by Semliki Forest and Chikungunya Viruses Is Dependent on nsP3 and Connected to Replication Complex Internalization. J. Virol. 2015, 89, 11420–11437. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; He, X.; Zhu, G.; Tu, H.; Liu, Z.; Li, W.; Han, S.; Yin, J.; Peng, B.; Liu, W. Coxsackievirus A16 elicits incomplete autophagy involving the mTOR and ERK pathways. PLoS ONE 2015, 10, e0122109. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.K.; Shrivastava, S.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J. Virol. 2012, 86, 6315–6322. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Panda, S.; Kudmulwar, D.; Chhatbar, S.P.; Nayak, S.C.; Krishnan, H.H. Hepatitis C virus NS5A binds to the mRNA cap-binding eukaryotic translation initiation 4F (eIF4F) complex and up-regulates host translation initiation machinery through eIF4E-binding protein 1 inactivation. J. Biol. Chem. 2012, 287, 5042–5058. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Nakao, H.; Tan, S.L.; Polyak, S.J.; Neddermann, P.; Vijaysri, S.; Jacobs, B.L.; Katze, M.G. Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with Grb2 and P85 phosphatidylinositol 3-kinase. J. Virol. 2002, 76, 9207–9217. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Liang, D.; Tong, W.; Li, J.; Yuan, Z. Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. J. Biol. Chem. 2010, 285, 20870–20881. [Google Scholar] [CrossRef] [PubMed]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The Hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Mohankumar, V.; Dhanushkodi, N.R.; Raju, R. Sindbis virus replication, is insensitive to rapamycin and torin1, and suppresses Akt/mTOR pathway late during infection in HEK cells. Biochem. Biophys. Res. Commun. 2011, 406, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Hardy, R.W. Role for the phosphatidylinositol 3-kinase-Akt-TOR pathway during sindbis virus replication in arthropods. J. Virol. 2012, 86, 3595–3604. [Google Scholar] [CrossRef] [PubMed]
- Shives, K.D.; Beatman, E.L.; Chamanian, M.; O’Brien, C.; Hobson-Peters, J.; Beckham, J.D. West nile virus-induced activation of mammalian target of rapamycin complex 1 supports viral growth and viral protein expression. J. Virol. 2014, 88, 9458–9471. [Google Scholar] [CrossRef] [PubMed]
- Urbanowski, M.D.; Hobman, T.C. The West Nile virus capsid protein blocks apoptosis through a phosphatidylinositol 3-kinase-dependent mechanism. J. Virol. 2013, 87, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Liao, C.L.; Lin, Y.L. Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J. Virol. 2005, 79, 8388–8399. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.N.; Gorbunova, E.E.; Mackow, E.R. Hypoxia induces permeability and giant cell responses of Andes virus-infected pulmonary endothelial cells by activating the mTOR-S6K signaling pathway. J. Virol. 2013, 87, 12999–13008. [Google Scholar] [CrossRef] [PubMed]
- McNulty, S.; Flint, M.; Nichol, S.T.; Spiropoulou, C.F. Host mTORC1 signaling regulates andes virus replication. J. Virol. 2013, 87, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Effect of the phosphatidylinositol 3-kinase/Akt pathway on influenza A virus propagation. J. Gen. Virol. 2007, 88, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Avota, E.; Avots, A.; Niewiesk, S.; Kane, L.P.; Bommhardt, U.; ter Meulen, V.; Schneider-Schaulies, S. Disruption of Akt kinase activation is important for immunosuppression induced by measles virus. Nat. Med. 2001, 7, 725–731. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.P.; de Freitas, D.N.; Antuntes Fernandes, K.E.; D’Avila da Cunha, M.; Antunes Fernandes, J.L.; Benetti Gassen, R.; Fazolo, T.; Pinto, L.A.; Scotta, M.; Mattiello, R.; et al. Respiratory syncytial virus induces phosphorylation of mTOR at ser2448 in CD8 T cells from nasal washes of infected infants. Clin. Exp. Immunol. 2016, 183, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Moy, R.H.; Gold, B.; Molleston, J.M.; Schad, V.; Yanger, K.; Salzano, M.V.; Yagi, Y.; Fitzgerald, K.A.; Stanger, B.Z.; Soldan, S.S.; et al. Antiviral autophagy restrictsRift Valley fever virus infection and is conserved from flies to mammals. Immunity 2014, 40, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Dunn, E.F.; Connor, J.H. Dominant inhibition of Akt/protein kinase B signaling by the matrix protein of a negative-strand RNA virus. J. Virol. 2011, 85, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar] [CrossRef] [PubMed]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Yamori, T. Phosphatidylinositol 3-kinase inhibitors: Promising drug candidates for cancer therapy. Cancer Sci. 2008, 99, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, C.C.; Choi, S.; McCormick, F.; Stokoe, D. Adenovirus overrides cellular checkpoints for protein translation. Cell Cycle 2005, 4, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Sala, C. PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 2001, 24, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.K.; Lee, S.S.; Thomas, D.L.; Latorre, I.J.; Weiss, R.S.; Glaunsinger, B.A.; Javier, R.T. Selective PDZ protein-dependent stimulation of phosphatidylinositol 3-kinase by the adenovirus E4-ORF1 oncoprotein. Oncogene 2003, 22, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Scholle, F.; Bendt, K.M.; Raab-Traub, N. Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J. Virol. 2000, 74, 10681–10689. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wan, X.; Jiang, R.; Deng, L.; Gao, Y.; Tang, J.; Yang, Y.; Zhao, W.; Yan, X.; Yao, K.; et al. Epstein-Barr virus-encoded latent membrane protein 2A promotes the epithelial-mesenchymal transition in nasopharyngeal carcinoma via metastatic tumor antigen 1 and mechanistic target of rapamycin signaling induction. J. Virol. 2014, 88, 11872–11885. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.; Pencil, S.D.; Nicolson, G.L. A novel candidate metastasis-associated gene, mta1, differentially expressed in highly metastatic mammary adenocarcinoma cell lines. cDNA cloning, expression, and protein analyses. J. Biol. Chem. 1994, 269, 22958–22963. [Google Scholar] [PubMed]
- Martin, D.; Nguyen, Q.; Molinolo, A.; Gutkind, J.S. Accumulation of dephosphorylated 4EBP after mTOR inhibition with rapamycin is sufficient to disrupt paracrine transformation by the KSHV vGPCR oncogene. Oncogene 2014, 33, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Stallone, G.; Schena, A.; Infante, B.; Di Paolo, S.; Loverre, A.; Maggio, G.; Ranieri, E.; Gesualdo, L.; Schena, F.P.; Grandaliano, G. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N. Engl. J. Med. 2005, 352, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Campistol, J.M.; Gutierrez-Dalmau, A.; Torregrosa, J.V. Conversion to sirolimus: a successful treatment for posttransplantation Kaposi’s sarcoma. Transplantation 2004, 77, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Beatman, E.; Oyer, R.; Shives, K.D.; Hedman, K.; Brault, A.C.; Tyler, K.L.; Beckham, J.D. West Nile virus growth is independent of autophagy activation. Virology 2012, 433, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, K.A.; Villarreal, L.P. Natural biology of polyomavirus middle T antigen. Microbiol. Mol. Biol. Rev. 2001, 65, 288–318, second and third pages, table of contents. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Veldman, T.; Rundell, K.; Schlegel, R. Simian virus 40 small tumor antigen activates AKT and telomerase and induces anchorage-independent growth of human epithelial cells. J. Virol. 2002, 76, 10685–10691. [Google Scholar] [CrossRef] [PubMed]
- Spuul, P.; Balistreri, G.; Kaariainen, L.; Ahola, T. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest Virus replication complexes from the plasma membrane to modified lysosomes. J. Virol. 2010, 84, 7543–7557. [Google Scholar] [CrossRef] [PubMed]
- Abere, B.; Wikan, N.; Ubol, S.; Auewarakul, P.; Paemanee, A.; Kittisenachai, S.; Roytrakul, S.; Smith, D.R. Proteomic analysis of chikungunya virus infected microgial cells. PLoS ONE 2012, 7, e34800. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, A.; Loiarro, M.; Bielli, P.; Busa, R.; Paronetto, M.P.; Loreni, F.; Geremia, R.; Sette, C. Phosphorylation of eIF4E by MNKs supports protein synthesis, cell cycle progression and proliferation in prostate cancer cells. Carcinogenesis 2008, 29, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Fesq, H.; Bacher, M.; Nain, M.; Gemsa, D. Programmed cell death (apoptosis) in human monocytes infected by influenza A virus. Immunobiology 1994, 190, 175–182. [Google Scholar] [CrossRef]
- Hinshaw, V.S.; Olsen, C.W.; Dybdahl-Sissoko, N.; Evans, D. Apoptosis: A mechanism of cell killing by influenza A and B viruses. J. Virol. 1994, 68, 3667–3673. [Google Scholar] [PubMed]
- Ehrhardt, C.; Marjuki, H.; Wolff, T.; Nurnberg, B.; Planz, O.; Pleschka, S.; Ludwig, S. Bivalent role of the phosphatidylinositol-3-kinase (PI3K) during influenza virus infection and host cell defence. Cell Microbiol. 2006, 8, 1336–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, X.; Liu, D.; Fan, Z.; Hu, X.; Yan, J.; Wang, M.; Gao, G.F. Autophagy is involved in influenza A virus replication. Autophagy 2009, 5, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Datan, E.; Shirazian, A.; Benjamin, S.; Matassov, D.; Tinari, A.; Malorni, W.; Lockshin, R.A.; Garcia-Sastre, A.; Zakeri, Z. mTOR/p70S6K signaling distinguishes routine, maintenance-level autophagy from autophagic cell death during influenza A infection. Virology 2014, 452–453, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Zhirnov, O.P.; Konakova, T.E.; Garten, W.; Klenk, H. Caspase-dependent N-terminal cleavage of influenza virus nucleocapsid protein in infected cells. J. Virol. 1999, 73, 10158–10163. [Google Scholar] [PubMed]
- Liu, G.; Zhong, M.; Guo, C.; Komatsu, M.; Xu, J.; Wang, Y.; Kitazato, K. Autophagy is involved in regulating influenza A virus RNA and protein synthesis associated with both modulation of Hsp90 induction and mTOR/p70S6K signaling pathway. Int. J. Biochem. Cell Biol. 2016, 72, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.A.; Leite, F.G.; Andrade, L.G.; Torres, A.A.; De Sousa, L.P.; Barcelos, L.S.; Teixeira, M.M.; Ferreira, P.C.; Kroon, E.G.; Souto-Padron, T.; et al. Activation of the PI3K/Akt pathway early during vaccinia and cowpox virus infections is required for both host survival and viral replication. J. Virol. 2009, 83, 6883–6899. [Google Scholar] [CrossRef] [PubMed]
- Allan, G.M.; McNeilly, F.; Cassidy, J.P.; Reilly, G.A.; Adair, B.; Ellis, W.A.; McNulty, M.S. Pathogenesis of porcine circovirus; experimental infections of colostrum deprived piglets and examination of pig foetal material. Vet. Microbiol. 1995, 44, 49–64. [Google Scholar] [CrossRef]
- Zhu, B.; Xu, F.; Li, J.; Shuai, J.; Li, X.; Fang, W. Porcine circovirus type 2 explores the autophagic machinery for replication in PK-15 cells. Virus Res. 2012, 163, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zhou, Y.; Xu, F.; Shuai, J.; Li, X.; Fang, W. Porcine circovirus type 2 induces autophagy via the AMPK/ERK/TSC2/mTOR signaling pathway in PK-15 cells. J. Virol. 2012, 86, 12003–12012. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J. Virol. 2011, 85, 9369–9376. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Del Prete, G.Q.; Maguire, T.G.; Alwine, J.C. AMPK-mediated inhibition of mTOR kinase is circumvented during immediate-early times of human cytomegalovirus infection. J. Virol. 2007, 81, 3649–3651. [Google Scholar] [CrossRef] [PubMed]
- Tilton, C.; Clippinger, A.J.; Maguire, T.; Alwine, J.C. Human cytomegalovirus induces multiple means to combat reactive oxygen species. J. Virol. 2011, 85, 12585–12593. [Google Scholar] [CrossRef] [PubMed]
- Werden, S.J.; McFadden, G. Pharmacological manipulation of the akt signaling pathway regulates myxoma virus replication and tropism in human cancer cells. J. Virol. 2010, 84, 3287–3302. [Google Scholar] [CrossRef] [PubMed]
- Stanford, M.M.; Barrett, J.W.; Nazarian, S.H.; Werden, S.; McFadden, G. Oncolytic virotherapy synergism with signaling inhibitors: Rapamycin increases myxoma virus tropism for human tumor cells. J. Virol. 2007, 81, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.H.; Lyles, D.S. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 2002, 76, 10177–10187. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, K.C.; Tartell, M.A.; Herrmann, C.; Hackett, B.A.; Taschuk, F.; Panda, D.; Menghani, S.V.; Sabin, L.R.; Cherry, S. Virus-induced translational arrest through 4EBP1/2-dependent decay of 5'-TOP mRNAs restricts viral infection. Proc. Natl. Acad. Sci. USA 2015, 112, E2920–E2929. [Google Scholar] [CrossRef] [PubMed]
- Carsillo, M.; Kim, D.; Niewiesk, S. Role of AKT kinase in measles virus replication. J. Virol. 2010, 84, 2180–2183. [Google Scholar] [CrossRef] [PubMed]
- Bueno, S.M.; Gonzalez, P.A.; Pacheco, R.; Leiva, E.D.; Cautivo, K.M.; Tobar, H.E.; Mora, J.E.; Prado, C.E.; Zuniga, J.P.; Jimenez, J.; et al. Host immunity during RSV pathogenesis. Int. Immunopharmacol. 2008, 8, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.L.; McDonald, N.J.; Sheng, J.; Shaw, M.W.; Hodge, T.W.; Rubin, D.H.; O’Brien, W.A.; Smee, D.F. Inhibition of influenza A virus replication by antagonism of a PI3K-AKT-mTOR pathway member identified by gene-trap insertional mutagenesis. Antivir. Chem. Chemother. 2012, 22, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003, 5, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, J.; Ling, M.T.; Zhao, L.; Zhao, K.N. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol. Cancer 2015, 14, 87. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Munger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef] [PubMed]
- Terhune, S.; Torigoi, E.; Moorman, N.; Silva, M.; Qian, Z.; Shenk, T.; Yu, D. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 2007, 81, 3109–3123. [Google Scholar] [CrossRef] [PubMed]
- Xuan, B.; Qian, Z.; Torigoi, E.; Yu, D. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 2009, 83, 3463–3474. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Xuan, B.; Liu, H.; Zhong, J.; Yu, D.; Qian, Z. Tuberous Sclerosis Complex Protein 2-Independent Activation of mTORC1 by Human Cytomegalovirus pUL38. J. Virol. 2015, 89, 7625–7635. [Google Scholar] [CrossRef] [PubMed]
- Rauwel, B.; Jang, S.M.; Cassano, M.; Kapopoulou, A.; Barde, I.; Trono, D. Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Benetti, L.; Roizman, B. Protein kinase B/Akt is present in activated form throughout the entire replicative cycle of deltaU(S)3 mutant virus but only at early times after infection with wild-type herpes simplex virus 1. J. Virol. 2006, 80, 3341–3348. [Google Scholar] [CrossRef] [PubMed]
- Chuluunbaatar, U.; Mohr, I. A herpesvirus kinase that masquerades as Akt: You don’t have to look like Akt, to act like it. Cell Cycle 2011, 10, 2064–2068. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Kennedy, S.G.; O’Leary, M.A.; Sonenberg, N.; Hay, N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998, 12, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Montero, H.; Garcia-Roman, R.; Mora, S.I. eIF4E as a control target for viruses. Viruses 2015, 7, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J. Virol. 2004, 78, 11030–11039. [Google Scholar] [CrossRef] [PubMed]
- Roizman, B.; Knipe, D. Herpes Simplex Viruses and Their Replication, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 2. [Google Scholar]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Chang, Y.; Moore, P.S. Merkel cell polyomavirus-positive Merkel cell carcinoma requires viral small T-antigen for cell proliferation. J. Investig. Dermatol. 2014, 134, 1479–1481. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Velasquez, C.; Cheng, E.; Cordek, D.G.; Kwun, H.J.; Chang, Y.; Moore, P.S. CDK1 substitutes for mTOR kinase to activate mitotic cap-dependent protein translation. Proc. Natl. Acad. Sci. USA 2015, 112, 5875–5882. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Kudchodkar, S.B.; Alwine, J.C. Effects of simian virus 40 large and small tumor antigens on mammalian target of rapamycin signaling: Small tumor antigen mediates hypophosphorylation of eIF4E-binding protein 1 late in infection. J. Virol. 2005, 79, 6882–6889. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Alwine, J.C. 19S late mRNAs of simian virus 40 have an internal ribosome entry site upstream of the virion structural protein 3 coding sequence. J. Virol. 2006, 80, 6553–6558. [Google Scholar] [CrossRef] [PubMed]
- Mannova, P.; Beretta, L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: Control of cell survival and viral replication. J. Virol. 2005, 79, 8742–8749. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Vedagiri, D.; Viveka, T.S.; Harshan, K.H. A unique phosphorylation-dependent eIF4E assembly on 40S ribosomes co-ordinated by hepatitis C virus protein NS5A that activates internal ribosome entry site translation. Biochem. J. 2014, 462, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Kang, R.; Wang, J.; Luo, G.; Yang, W.; Zhao, Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013, 9, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Chisari, F.V. Autophagy proteins promote hepatitis C virus replication. Autophagy 2009, 5, 1224–1225. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Vandergaast, R.; Fredericksen, B.L. West Nile virus (WNV) replication is independent of autophagy in mammalian cells. PLoS ONE 2012, 7, e45800. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Castillo, C.; Naranjo, J.; Sepulveda, A.; Ossa, G.; Levy, H. Hantavirus pulmonary syndrome due to Andes virus in Temuco, Chile: Clinical experience with 16 adults. Chest 2001, 120, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.; Padula, P.; Rossi, C.; Lazaro, M.E.; Franze-Fernandez, M.T. Genetic identification of a new hantavirus causing severe pulmonary syndrome in Argentina. Virology 1996, 220, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Daussy, C.F.; Beaumelle, B.; Espert, L. Autophagy restricts HIV-1 infection. Oncotarget 2015, 6, 20752–20753. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, C.; Pilli, M.; Kehrl, J.H. Roles of autophagy in HIV infection. Immunol. Cell Biol. 2015, 93, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy through Transcription Factor EB Sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [PubMed]
- Cinti, A.; Le Sage, V.; Milev, M.P.; Crossie, C.; Valiente-Echeverría, F.; Olivier, F.; Mouland, A.J. HIV-1 enhances mTORC1 activity and repositions lysosomes to the periphery by co-opting Rag GTPases. Manuscript in preparation.
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; Del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; McCubrey, J.A.; Bendtzen, K.; Nicoletti, F. Potential use of rapamycin in HIV infection. Br. J. Clin. Pharmacol. 2010, 70, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Group Classification | Virus | Viral Protein | Target | Reference |
|---|---|---|---|---|
| dsDNA | Adenovirus | E4orf1 | PI3K activation | [19,20,21] |
| dsDNA | Adenovirus | E4orf4 | Blocks dephosphorylation of mTORC1 via PP2A | [22] |
| dsDNA | Epstein-Barr virus | LMP2A | PI3K activation | [23] |
| dsDNA | Human cytomegalovirus | IEP72 and IEP86 | Activates Akt | [24] |
| dsDNA | Human cytomegalovirus | N/D | Increase in abundance of eIF4F complex proteins | [25] |
| dsDNA | Human cytomegalovirus | UL38 | Binds and antagonizes TSC2 | [26] |
| dsDNA | Human cytomegalovirus | N/D | Redistribution of mTORC1 to a perinuclear localization | [27] |
| dsDNA | Human papillomavirus | protein E6 | Causes degradation of TSC2 | [28,29] |
| dsDNA | Human papillomavirus | protein E7 | Inhibits dephosphorylation of Akt through an interaction with PP2A | [30] |
| dsDNA | Herpes simplex virus type 1 | Us3 | Akt mimic | [31] |
| dsDNA | Herpes simplex virus type 1 | ICP0 | Degradation of 4EBP1 by the proteasome | [32] |
| dsDNA | Herpes simplex virus type 1 | ICP6 | Associates with eIF4G | [33] |
| dsDNA | Kaposi’s Sarcoma Herpesvirus | vGPCR | PI3K activation | [34,35] |
| dsDNA | Merkel cell polyomavirus | sT | Hyperphosphorylation of 4EBP1 | [36] |
| dsDNA | Murine polyomavirus | MT | PI3K activation | [37,38] |
| dsDNA | Myxoma virus | M-T5 | Activates Akt | [39,40] |
| dsDNA | Simian virus 40 | sT | Activates Akt through an interaction with PP2A | [41,42] |
| dsDNA | Vaccinia virus | N/D | Mediates activation of PI3K/Akt through protein integrin β1 (ITGβ1) | [43] |
| dsDNA | Vaccinia virus | N/D | Alters architecture of eIF4F complex | [44] |
| dsDNA | Varicella zoster virus | pORFs 47 and 66 | Activates Akt | [45] |
| ssDNA | Porcine circovirus type 2 | N/D | Activates PI3K | [46] |
| dsRNA | Avian reovirus | Protein p17 | Inactivation of Akt through activation of PTEN | [47] |
| dsRNA | Infectious bursal disease virus | VP2 capsid | Inactivates Akt | [48] |
| +ssRNA | Chikungunya virus | N/D | Controversial activation or suppression of PI3K/Akt/mTOR pathway | [49,50,51,52] |
| +ssRNA | Coxsackievirus A16 | N/D | Inhibits Akt phosphorylation | [53] |
| +ssRNA | Hepatitis C virus | NS5A | Activation of PI3K/Akt/mTOR pathway | [54,55,56,57,58] |
| +ssRNA | Human immunodeficiency virus type 1 | Env | Activation of mTOR | [59] |
| +ssRNA | Semliki Forest virus | nsP3 | Activation of Akt | [52] |
| +ssRNA | Sindbis virus | N/D | Suppression in HEK cells and activation in arthropod cells of PI3K/Akt/mTOR pathway | [60,61] |
| +ssRNA | West Nile virus, Dengue virus, Japanese encephalitis virus | N/D | PI3K activation | [62,63,64] |
| −ssRNA | Andes virus | Gn | Modulation of mTOR and lysosomal signaling | [65,66] |
| −ssRNA | Influenza A virus | NS1 | Activates PI3K | [67,68] |
| −ssRNA | Measles virus | N/D | Inactivates Akt | [69] |
| −ssRNA | Respiratory syncytial virus | F protein | Induces phosphorylation of mTOR via a PI3K-independent mechanism | [70] |
| −ssRNA | Rift Valley virus | N/D | Inhibits Akt phosphorylation | [71] |
| −ssRNA | Vesicular stomatitis virus | Matrix protein M | Inactivates Akt | [72] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A.J. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses 2016, 8, 152. https://doi.org/10.3390/v8060152
Le Sage V, Cinti A, Amorim R, Mouland AJ. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses. 2016; 8(6):152. https://doi.org/10.3390/v8060152
Chicago/Turabian StyleLe Sage, Valerie, Alessandro Cinti, Raquel Amorim, and Andrew J. Mouland. 2016. "Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway" Viruses 8, no. 6: 152. https://doi.org/10.3390/v8060152
APA StyleLe Sage, V., Cinti, A., Amorim, R., & Mouland, A. J. (2016). Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses, 8(6), 152. https://doi.org/10.3390/v8060152