
Phleboviruses and the Type I Interferon Response


Abstract
:1. Introduction
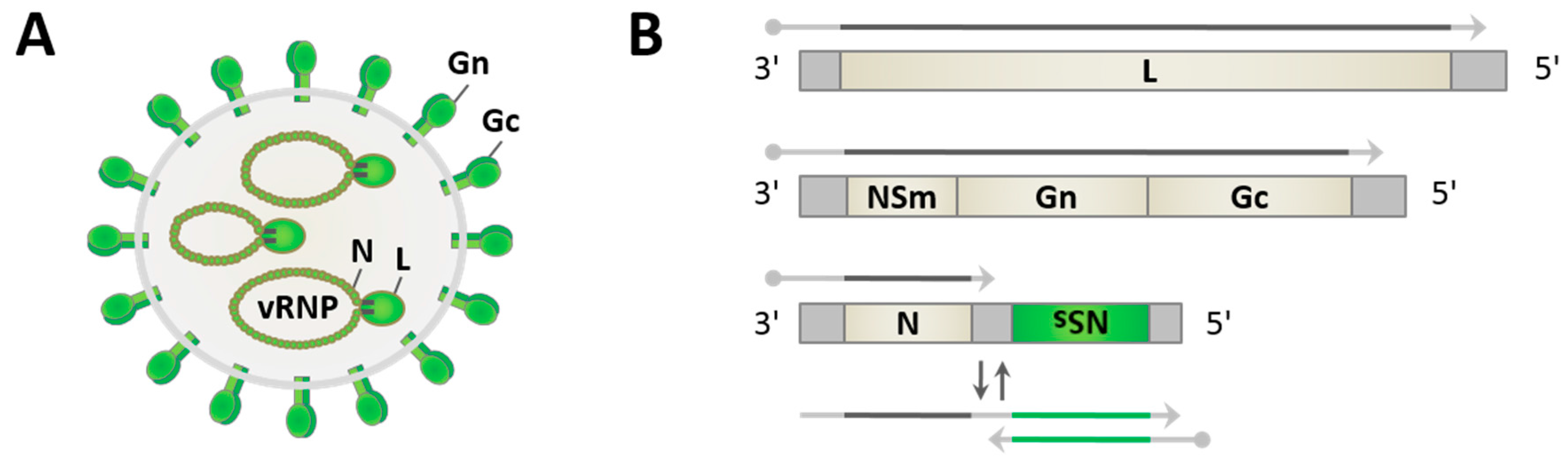
2. Phleboviruses—An Emerging Group of Arthropod-Transmitted Pathogens
3. Viral Replication in the Mammalian Host
4. The Type I Interferon System in RNA Virus Infection
5. Activation of the Interferon System by Phleboviruses
6. Viral Countermeasures
7. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Walter, C.T.; Barr, J.N. Recent advances in the molecular and cellular biology of bunyaviruses. J. Gen. Virol. 2011, 92, 2467–2484. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses. Phleboviruses. Available online: http://ictvonline.org/virusTaxonomy.asp?version=2012 (accessed on 15 July 2012).
- Alkan, C.; Alwassouf, S.; Piorkowski, G.; Bichaud, L.; Tezcan, S.; Dincer, E.; Ergunay, K.; Ozbel, Y.; Alten, B.; de Lamballerie, X.; et al. Isolation, genetic characterization, and seroprevalence of Adana virus, a novel phlebovirus belonging to the Salehabad virus complex, in Turkey. J. Virol. 2015, 89, 4080–4091. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Kasap, O.E.; Alten, B.; de Lamballerie, X.; Charrel, R.N. Sandfly-borne phlebovirus isolations from Turkey: New insight into the sandfly fever Sicilian and sandfly fever Naples species. PLoS Negl. Trop. Dis. 2016, 10, e0004519. [Google Scholar] [CrossRef] [PubMed]
- Amaro, F.; Hanke, D.; Ze-Ze, L.; Alves, M.J.; Becker, S.C.; Hoper, D. Genetic characterization of Arrabida virus, a novel phlebovirus isolated in South Portugal. Virus Res. 2016, 214, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Amaro, F.; Ze-Ze, L.; Alves, M.J.; Borstler, J.; Clos, J.; Lorenzen, S.; Becker, S.C.; Schmidt-Chanasit, J.; Cadar, D. Co-circulation of a novel phlebovirus and Massilia virus in sandflies, Portugal. Virol. J. 2015, 12, 174. [Google Scholar] [CrossRef] [PubMed]
- Bichaud, L.; Dachraoui, K.; Alwassouf, S.; Alkan, C.; Mensi, M.; Piorkowski, G.; Sakhria, S.; Seston, M.; Fares, W.; de Lamballerie, X.; et al. Isolation, full genomic characterization and neutralization-based human seroprevalence of Medjerda Valley virus, a novel sandfly-borne phlebovirus belonging to the Salehabad virus complex in northern Tunisia. J. Gen. Virol. 2016, 97, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Charrel, R.N.; Moureau, G.; Temmam, S.; Izri, A.; Marty, P.; Parola, P.; da Rosa, A.T.; Tesh, R.B.; de Lamballerie, X. Massilia virus, a novel Phlebovirus (Bunyaviridae) isolated from sandflies in the Mediterranean. Vector Borne Zoonotic Dis. 2009, 9, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Dachraoui, K.; Fares, W.; Bichaud, L.; Barhoumi, W.; Beier, J.C.; Derbali, M.; Cherni, S.; Lamballerie, X.; Chelbi, I.; Charrel, R.N.; et al. Phleboviruses associated with sand flies in arid bio-geographical areas of Central Tunisia. Acta Trop. 2016, 158, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Weisend, C.; Travassos da Rosa, A.P.; Anzick, S.L.; Dahlstrom, E.; Porcella, S.F.; Dorward, D.W.; Yu, X.J.; Tesh, R.B.; Ebihara, H. Characterization of the Bhanja serogroup viruses (Bunyaviridae): A novel species of the genus Phlebovirus and its relationship with other emerging tick-borne phleboviruses. J. Virol. 2013, 87, 3719–3728. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Weisend, C.; Kajihara, M.; Matysiak, C.; Williamson, B.N.; Simuunza, M.; Mweene, A.S.; Takada, A.; Tesh, R.B.; Ebihara, H. Comprehensive molecular detection of tick-borne phleboviruses leads to the retrospective identification of taxonomically unassigned bunyaviruses and the discovery of a novel member of the genus phlebovirus. J. Virol. 2015, 89, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; da Rosa, A.T.; Savji, N.; Sze, W.; Wick, I.; Guzman, H.; Hutchison, S.; Tesh, R.; Lipkin, W.I. Aguacate virus, a new antigenic complex of the genus Phlebovirus (family Bunyaviridae). J. Gen. Virol. 2011, 92, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Tesh, R.; Travassos da Rosa, A.; Savji, N.; Sze, W.; Jain, K.; Serge, R.; Guzman, H.; Guevara, C.; Nunes, M.R.; et al. Characterization of the Candiru antigenic complex (Bunyaviridae: Phlebovirus), a highly diverse and reassorting group of viruses affecting humans in tropical America. J. Virol. 2011, 85, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Savji, N.; Travassos da Rosa, A.; Desai, A.; Sanchez-Seco, M.P.; Guzman, H.; Lipkin, W.I.; Tesh, R. Characterization of the Salehabad virus species complex of the genus Phlebovirus (Bunyaviridae). J. Gen. Virol. 2013, 94, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Savji, N.; Travassos da Rosa, A.; Guzman, H.; Yu, X.; Desai, A.; Rosen, G.E.; Hutchison, S.; Lipkin, W.I.; et al. Characterization of the Uukuniemi virus group (Phlebovirus: Bunyaviridae): Evidence for seven distinct species. J. Virol. 2013, 87, 3187–3195. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Tesh, R.B.; Savji, N.; Travassos da Rosa, A.P.; Guzman, H.; Bussetti, A.V.; Desai, A.; Ladner, J.; Sanchez-Seco, M.; Lipkin, W.I. Characterization of the sandfly fever Naples species complex and description of a new Karimabad species complex (genus Phlebovirus, family Bunyaviridae). J. Gen. Virol. 2014, 95, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Wiley, M.R.; Travassos da Rosa, A.P.; Guzman, H.; Quiroz, E.; Savji, N.; Carrera, J.P.; Bussetti, A.V.; Ladner, J.T.; Lipkin, W.I.; et al. Characterization of the Punta Toro species complex (genus Phlebovirus, family Bunyaviridae). J. Gen. Virol. 2015, 96, 2079–2085. [Google Scholar] [CrossRef] [PubMed]
- Remoli, M.E.; Fortuna, C.; Marchi, A.; Bucci, P.; Argentini, C.; Bongiorno, G.; Maroli, M.; Gradoni, L.; Gramiccia, M.; Ciufolini, M.G. Viral isolates of a novel putative phlebovirus in the Marche Region of Italy. Am. J. Trop. Med. Hyg. 2014, 90, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Chen, H.; Travassos, A.P.; Tesh, R.B.; Xiao, S.Y. Phylogenetic relationships among sandfly fever group viruses (Phlebovirus: Bunyaviridae) based on the small genome segment. J. Gen. Virol. 2007, 88, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.Y.; Krishnamurthy, S.; Cai, Z.Q.; Popov, V.L.; da Rosa, A.P.T.; Guzman, H.; Cao, S.; Virgin, H.W.; Tesh, R.B.; Wang, D. Identification of novel viruses using VirusHunter—An automated data analysis pipeline. PLoS ONE 2013, 8, e78470. [Google Scholar] [CrossRef] [PubMed]
- Zhioua, E.; Moureau, G.; Chelbi, I.; Ninove, L.; Bichaud, L.; Derbali, M.; Champs, M.; Cherni, S.; Salez, N.; Cook, S.; et al. Punique virus, a novel phlebovirus, related to sandfly fever Naples virus, isolated from sandflies collected in Tunisia. J. Gen. Virol. 2010, 91, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Bichaud, L.; de Lamballerie, X.; Alten, B.; Gould, E.A.; Charrel, R.N. Sandfly-borne phleboviruses of Eurasia and Africa: Epidemiology, genetic diversity, geographic range, control measures. Antivir. Res. 2013, 100, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M.; Brennan, B. Emerging phleboviruses. Curr. Opin. Virol. 2014, 5, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Boshra, H.; Lorenzo, G.; Busquets, N.; Brun, A. Rift Valley fever: Recent insights into pathogenesis and prevention. J. Virol. 2011, 85, 6098–6105. [Google Scholar] [CrossRef] [PubMed]
- Bird, B.H.; Ksiazek, T.G.; Nichol, S.T.; Maclachlan, N.J. Rift Valley fever virus. J. Am. Vet. Med. Assoc. 2009, 234, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Daubney, R.; Hudson, J.R.; Garnham, P.C. Enzootic hepatitis or Rift Valley fever. An undescribed virus disease of sheep cattle and man from East Africa. J. Pathol. Bacteriol. 1931, 34, 545–579. [Google Scholar] [CrossRef]
- Arishi, H.; Ageel, A.; Rahman, M.A.; Hazmi, A.A.; Arishi, A.R.; Ayoola, B.; Menon, C.; Ashraf, J.; Frogusin, O.; Sawwan, F.; et al. Outbreak of Rift Valley fever—Saudi Arabia, August-October, 2000. MMMW Rep. 2000, 49, 905–908. [Google Scholar]
- Charrel, R.N.; Bichaud, L.; de Lamballerie, X. Emergence of Toscana virus in the mediterranean area. World J. Virol. 2012, 1, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Srihongse, S.; Johnson, C.M. Human infections with Chagres virus in Panama. Am. J. Trop. Med. Hyg. 1974, 23, 690–693. [Google Scholar] [PubMed]
- Travassos da Rosa, A.P.; Tesh, R.B.; Pinheiro, F.P.; Travassos da Rosa, J.F.; Peterson, N.E. Characterization of eight new phlebotomus fever serogroup arboviruses (Bunyaviridae: Phlebovirus) from the Amazon region of Brazil. Am. J. Trop. Med. Hyg. 1983, 32, 1164–1171. [Google Scholar] [PubMed]
- Tesh, R.B. The genus Phlebovirus and its vectors. Annu. Rev. Entomol. 1988, 33, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Rolin, A.I.; Berrang-Ford, L.; Kulkarni, M.A. The risk of Rift Valley fever virus introduction and establishment in the United States and European Union. Emerg. Microbes Infect. 2013, 2, e81. [Google Scholar] [CrossRef] [PubMed]
- Salman, M. Is the United States really at risk for introduction of Rift Valley fever virus? J. Am. Vet. Med. Assoc. 2013, 242, 606–608. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, L.; Du, Y.; Ma, H.; Mu, Y.; Tang, X.; Wang, H.; Kang, K.; Zhang, S.; Wu, W.; et al. Detection of a novel bunyavirus associated with fever, thrombocytopenia and leukopenia syndrome in Henan Province, China, using real-time reverse transcription PCR. J. Med. Microbiol. 2013, 62, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.M.; Zhao, L.; Wen, H.L.; Zhang, Z.T.; Liu, J.W.; Fang, L.Z.; Xue, Z.F.; Ma, D.Q.; Zhang, X.S.; Ding, S.J.; et al. Haemaphysalis longicornis ticks as reservoir and vector of severe fever with thrombocytopenia syndrome virus in China. Emerg. Infect. Dis. 2015, 21, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Liu, L.; Huang, X.; Ma, H.; Zhang, Y.; Du, Y.; Wang, P.; Tang, X.; Wang, H.; Kang, K.; et al. Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan Province, China: Discovery of a new bunyavirus. PLoS Pathog. 2011, 7, e1002369. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.J.; Liang, M.F.; Zhang, S.Y.; Liu, Y.; Li, J.D.; Sun, Y.L.; Zhang, L.; Zhang, Q.F.; Popov, V.L.; Li, C.; et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 2011, 364, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Zhou, D.J.; Qin, X.C.; Tian, J.H.; Xiong, Y.; Wang, J.B.; Chen, X.P.; Gao, D.Y.; He, Y.W.; Jin, D.; et al. The ecology, genetic diversity, and phylogeny of Huaiyangshan virus in China. J. Virol. 2012, 86, 2864–2868. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Yi, J.; Kim, G.; Choi, S.J.; Jun, K.I.; Kim, N.H.; Choe, P.G.; Kim, N.J.; Lee, J.K.; Oh, M.D. Severe fever with thrombocytopenia syndrome, South Korea, 2012. Emerg. Infect. Dis. 2013, 19, 1892–1894. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Han, M.G.; Yun, S.M.; Park, C.; Lee, W.J.; Ryou, J. Severe Fever with thrombocytopenia syndrome virus, South Korea, 2013. Emerg. Infect. Dis. 2014, 20, 1880–1882. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Maeda, K.; Suzuki, T.; Ishido, A.; Shigeoka, T.; Tominaga, T.; Kamei, T.; Honda, M.; Ninomiya, D.; Sakai, T.; et al. The First Identification and retrospective study of severe fever with thrombocytopenia syndrome in Japan. J. Infect. Dis. 2014, 209, 816–827. [Google Scholar] [CrossRef] [PubMed]
- McMullan, L.K.; Folk, S.M.; Kelly, A.J.; MacNeil, A.; Goldsmith, C.S.; Metcalfe, M.G.; Batten, B.C.; Albarino, C.G.; Zaki, S.R.; Rollin, P.E.; et al. A New phlebovirus associated with severe febrile illness in Missouri. N. Engl. J. Med. 2012, 367, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Savage, H.M.; Godsey, M.S.; Lambert, A.; Panella, N.A.; Burkhalter, K.L.; Harmon, J.R.; Lash, R.R.; Ashley, D.C.; Nicholson, W.L. First detection of Heartland virus (Bunyaviridae: Phlebovirus) from ffield collected arthropods. Am. J. Trop. Med. Hyg. 2013, 89, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Dilcher, M.; Alves, M.J.; Finkeisen, D.; Hufert, F.; Weidmann, M. Genetic characterization of Bhanja virus and Palma virus, two tick-borne phleboviruses. Virus Genes 2012, 45, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Carhan, A.; Uyar, Y.; Ozkaya, E.; Ertek, M.; Dobler, G.; Dilcher, M.; Wang, Y.; Spiegel, M.; Hufert, F.; Weidmann, M. Characterization of a sandfly fever Sicilian virus isolated during a sandfly fever epidemic in Turkey. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2010, 48, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Ergunay, K.; Ismayilova, V.; Colpak, I.A.; Kansu, T.; Us, D. A case of central nervous system infection due to a novel sandfly fever virus (SFV) variant: Sandfly fever Turkey virus (SFTV). J. Clin. Virol. 2012, 54, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Kocak Tufan, Z.; Weidmann, M.; Bulut, C.; Kinikli, S.; Hufert, F.T.; Dobler, G.; Demiroz, A.P. Clinical and laboratory findings of a sandfly fever Turkey virus outbreak in Ankara. J. Infect. 2011, 63, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Pardalos, G.; Athanasiou-Metaxa, M.; Papa, A. Novel phlebovirus in febrile child, Greece. Emerg. Infect. Dis. 2011, 17, 940–941. [Google Scholar] [CrossRef] [PubMed]
- Collao, X.; Palacios, G.; de Ory, F.; Sanbonmatsu, S.; Perez-Ruiz, M.; Navarro, J.M.; Molina, R.; Hutchison, S.K.; Lipkin, W.I.; Tenorio, A.; et al. Granada Virus: A natural phlebovirus reassortant of the sandfly fever Naples serocomplex with low seroprevalence in humans. Am. J. Trop. Med. Hyg. 2010, 83, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Huiskonen, J.T.; Overby, A.K.; Weber, F.; Grunewald, K. Electron cryo-microscopy and single-particle averaging of Rift Valley fever virus: Evidence for Gn-Gc glycoprotein heterodimers. J. Virol. 2009, 83, 3762–3769. [Google Scholar] [CrossRef] [PubMed]
- Overby, A.K.; Pettersson, R.F.; Grunewald, K.; Huiskonen, J.T. Insights into bunyavirus architecture from electron cryotomography of Uukuniemi virus. Proc. Natl. Acad. Sci. USA 2008, 105, 2375–2379. [Google Scholar] [CrossRef] [PubMed]
- Albarino, C.G.; Bird, B.H.; Nichol, S.T. A shared transcription termination signal on negative and ambisense RNA genome segments of Rift Valley fever, sandfly fever Sicilian, and Toscana viruses. J. Virol. 2007, 81, 5246–5256. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Won, S.; Peters, C.J.; Makino, S. Characterization of Rift Valley fever virus transcriptional terminations. J. Virol. 2007, 81, 8421–8438. [Google Scholar] [CrossRef] [PubMed]
- Lara, E.; Billecocq, A.; Leger, P.; Bouloy, M. Characterization of wild-type and alternate transcription termination signals in the Rift Valley fever virus genome. J. Virol. 2011, 85, 12134–12145. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, M.J.; Pettersson, R.F.; Baltimore, D. Circular forms of Uukuniemi virion RNA: an electron microscopic study. J. Virol. 1977, 21, 1085–1093. [Google Scholar] [PubMed]
- Leger, P.; Lozach, P.Y. Bunyaviruses: From transmission by arthropods to virus entry into the mammalian host first-target cells. Future Virol. 2015, 10, 859–881. [Google Scholar] [CrossRef]
- Lozach, P.Y.; Mancini, R.; Bitto, D.; Meier, R.; Oestereich, L.; Overby, A.K.; Pettersson, R.F.; Helenius, A. Entry of bunyaviruses into mammalian cells. Cell Host Microbe 2010, 7, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Lozach, P.Y.; Kuhbacher, A.; Meier, R.; Mancini, R.; Bitto, D.; Bouloy, M.; Helenius, A. DC-SIGN as a receptor for phleboviruses. Cell Host Microbe 2011, 10, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Klemm, C.; Reguera, J.; Cusack, S.; Zielecki, F.; Kochs, G.; Weber, F. Systems To establish bunyavirus genome replication in the absence of transcription. J. Virol. 2013, 87, 8205–8212. [Google Scholar] [CrossRef] [PubMed]
- Morin, B.; Coutard, B.; Lelke, M.; Ferron, F.; Kerber, R.; Jamal, S.; Frangeul, A.; Baronti, C.; Charrel, R.; de Lamballerie, X.; et al. The N-terminal domain of the Arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathog. 2010, 6, 8990–8995. [Google Scholar] [CrossRef] [PubMed]
- Bird, B.H.; Albarino, C.G.; Hartman, A.L.; Erickson, B.R.; Ksiazek, T.G.; Nichol, S.T. Rift Valley fever virus lacking the NSs and NSm genes is highly attenuated, confers protective immunity from virulent virus challenge, and allows for differential identification of infected and vaccinated animals. J. Virol. 2008, 82, 2681–2691. [Google Scholar] [CrossRef] [PubMed]
- Bird, B.H.; Maartens, L.H.; Campbell, S.; Erasmus, B.J.; Erickson, B.R.; Dodd, K.A.; Spiropoulou, C.F.; Cannon, D.; Drew, C.P.; Knust, B.; et al. Rift Valley fever virus vaccine lacking the NSs and NSm genes is safe, nonteratogenic, and confers protection from viremia, pyrexia, and abortion following challenge in adult and pregnant sheep. J. Virol. 2011, 85, 12901–12909. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, S.R.; Bird, B.H.; Albarino, C.G.; Nichol, S.T. The NSm proteins of Rift Valley fever virus are dispensable for maturation, replication and infection. Virology 2007, 359, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Ikegami, T.; Peters, C.J.; Makino, S. NSm and 78-kilodalton proteins of Rift Valley fever virus are nonessential for viral replication in cell culture. J. Virol. 2006, 80, 8274–8278. [Google Scholar] [CrossRef] [PubMed]
- Won, S.Y.; Ikegami, T.; Peters, C.J.; Makino, S. NSm protein of Rift Valley fever virus suppresses virus-induced apoptosis. J. Virol. 2007, 81, 13335–13345. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Weber, F. Segmented negative-strand RNA viruses and RIG-I: Divide (your genome) and rule. Curr. Opin. Microbiol. 2014, 20, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Weber, F. RIG-I-like receptors and negative-strand RNA viruses: RLRly bird catches some worms. Cytokine Growth Factor Rev. 2014, 25, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.T.; Mendoza, J.L.; Garcia, K.C.; Oldstone, M.B. Alpha and beta type 1 interferon signaling: Passage for diverse biologic outcomes. Cell 2016, 164, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Sousa, C.R.E. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M. Master sensors of pathogenic RNA—RIG-I like receptors. Immunobiology 2013, 218, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Sparrer, K.M.; Gack, M.U. Intracellular detection of viral nucleic acids. Curr. Opin. Microbiol. 2015, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Gack, M.U. Ubiquitination in the antiviral immune response. Virology 2015, 479–480, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, J.; Cai, X.; Wu, J.; Chen, X.; Wu, Y.T.; Sun, L.; Chen, Z.J. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife 2013, 2, e00785. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; Sousa, C.R.E. Cytosolic Sensing of Viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.D. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 2012, 12, 367–82. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. MMBR 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Garcia, M.A.; Gomez-Puertas, P.; Guerra, S.; Rullas, J.; Nakano, H.; Alcami, J.; Esteban, M. TRAF family proteins link PKR with NF-kappa B activation. Mol. Cell. Biol. 2004, 24, 4502–4512. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Sen, G.C. Interferon-induced IFIT Proteins: Their role in viral pathogenesis. J. Virol. 2015, 89, 2462–2468. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Marie, I.; Smith, E.; Prakash, A. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J. Interferon Cytokine Res. 2002, 22, 87–93. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [PubMed]
- Zielecki, F.; Weber, M.; Eickmann, M.; Spiegelberg, L.; Zaki, A.M.; Matrosovich, M.; Becker, S.; Weber, F. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J. Virol. 2013, 87, 5300–5304. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Andersson, I.; Klingstrom, J.; Schumann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Muhlberger, E.; et al. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Gawanbacht, A.; Habjan, M.; Rang, A.; Bomer, C.; Schmidt, A.M.; Veitinger, S.; Jacob, R.; Devignot, S.; Kochs, G.; et al. Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe 2013, 13, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Ermler, M.E.; Yerukhim, E.; Schriewer, J.; Schattgen, S.; Traylor, Z.; Wespiser, A.R.; Caffrey, D.R.; Chen, Z.J.J.; King, C.H.; Gale, M.; et al. RNA helicase signaling is critical for type I interferon production and protection against Rift Valley fever virus during mucosal challenge. J. Virol. 2013, 87, 4846–4860. [Google Scholar] [CrossRef] [PubMed]
- Gowen, B.B.; Hoopes, J.D.; Wong, M.H.; Jung, K.H.; Isakson, K.C.; Alexopoulou, L.; Flavell, R.A.; Sidwell, R.W. TLR3 deletion limits mortality and disease severity due to phlebovirus infection. J. Immunol. 2006, 177, 6301–6307. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Pichlmair, A.; Elliott, R.M.; Overby, A.K.; Glatter, T.; Gstaiger, M.; Superti-Furga, G.; Unger, H.; Weber, F. NSs protein of Rift Valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J. Virol. 2009, 83, 4365–4375. [Google Scholar] [CrossRef] [PubMed]
- Kende, M. Prophylactic and therapeutic efficacy of poly(I,C)-LC against Rift-Valley fever virus-infection in Mice. J. Biol. Response Modif. 1985, 4, 503–511. [Google Scholar]
- Peters, C.J.; Reynolds, J.A.; Slone, T.W.; Jones, D.E.; Stephen, E.L. Prophylaxis of Rift Valley fever with antiviral drugs, immune serum, an interferon inducer, and a macrophage activator. Antivir. Res. 1986, 6, 285–297. [Google Scholar] [CrossRef]
- Gowen, B.B.; Wong, M.H.; Jung, K.H.; Sanders, A.B.; Mitchell, W.M.; Alexopoulou, L.; Flavell, R.A.; Sidwell, R.W. TLR3 is essential for the induction of protective immunity against Punta Toro virus infection by the double-stranded RNA (dsRNA), poly(I:C12U), but not poly(I:C): Differential, recognition of synthetic dsRNA molecules. J. Immunol. 2007, 178, 5200–5208. [Google Scholar] [CrossRef] [PubMed]
- Sidwell, R.W.; Huffman, J.H.; Smee, D.F.; Gilbert, J.; Gessaman, A.; Pease, A.; Warren, R.P.; Huggins, J.; Kende, M. Potential role of immunomodulators for treatment of phlebovirus infections of animals. Ann. N. Y. Acad. Sci. 1992, 653, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Sidwell, R.W.; Huffman, J.H.; Barnard, D.L.; Smee, D.F.; Warren, R.P.; Chirigos, M.A.; Kende, M.; Huggins, J. Antiviral and immunomodulating inhibitors of experimentally-induced Punta Toro virus infections. Antivir. Res. 1994, 25, 105–122. [Google Scholar] [CrossRef]
- Pifat, D.Y.; Smith, J.F. Punta Toro virus infection of C57BL/6J mice—A model for phlebovirus-induced disease. Microb Pathog. 1987, 3, 409–422. [Google Scholar] [CrossRef]
- Do Valle, T.Z.; Billecocq, A.; Guillemot, L.; Alberts, R.; Gommet, C.; Geffers, R.; Calabrese, K.; Schughart, K.; Bouloy, M.; Montagutelli, X.; et al. A new mouse model reveals a critical role for host innate immunity in resistance to Rift Valley fever. J. Immunol. 2010, 185, 6146–6156. [Google Scholar] [CrossRef] [PubMed]
- Morrill, J.C.; Jennings, G.B.; Johnson, A.J.; Cosgriff, T.M.; Gibbs, P.H.; Peters, C.J. Pathogenesis of Rift Valley fever in Rhesus monkeys—Role of interferon response. Arch. Virol. 1990, 110, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Bouloy, M.; Janzen, C.; Vialat, P.; Khun, H.; Pavlovic, J.; Huerre, M.; Haller, O. Genetic evidence for an interferon-antagonistic function of Rift Valley fever virus nonstructural protein NSs. J. Virol. 2001, 75, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, M.; Wong, M.H.; Skirpstunas, R.; Morrey, J.D.; Gowen, B.B. Punta Toro virus (Bunyaviridae, Phlebovirus) infection in mice: Strain differences in pathogenesis and host interferon response. Virology 2009, 395, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Frese, M.; Kochs, G.; Feldmann, H.; Hertkorn, C.; Haller, O. Inhibition of bunyaviruses, phleboviruses, and hantaviruses by human MxA protein. J. Virol. 1996, 70, 915–923. [Google Scholar] [PubMed]
- Sandrock, M.; Frese, M.; Haller, O.; Kochs, G. Interferon-induced rat Mx proteins confer resistance to Rift Valley fever virus and other arthropod-borne viruses. J. Interferon Cytokine Res. 2001, 21, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Penski, N.; Wagner, V.; Spiegel, M.; Overby, A.K.; Kochs, G.; Huiskonen, J.T.; Weber, F. Efficient production of Rift Valley fever virus-like particles: The antiviral protein MxA can inhibit primary transcription of bunyaviruses. Virology 2009, 385, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Janzen, C.; Hohenberg, H.; Haller, O. Antivirally active MxA protein sequesters La Crosse virus nucleocapsid protein into perinuclear complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 3153–3158. [Google Scholar] [CrossRef] [PubMed]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Radoshitzky, S.R.; Kota, K.P.; Altamura, L.A.; Smith, J.M.; Packard, B.Z.; Kuhn, J.H.; Costantino, J.; et al. IFITM-2 and IFITM-3 but not IFITM-1 restrict Rift Valley fever virus. J. Virol. 2013, 87, 8451–8464. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Narayanan, K.; Won, S.; Kamitani, W.; Peters, C.J.; Makino, S. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PLoS Pathog. 2009, 5, e1000287. [Google Scholar] [CrossRef] [PubMed]
- Atasheva, S.; Akhrymuk, M.; Frolova, E.I.; Frolov, I. New PARP gene with an anti-alphavirus function. J. Virol. 2012, 86, 8147–8160. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Lassnig, C.; Eberle, C.A.; Gorna, M.W.; Baumann, C.L.; Burkard, T.R.; Burckstummer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Vialat, P.; Billecocq, A.; Kohl, A.; Bouloy, M. The S segment of Rift Valley fever phlebovirus (Bunyaviridae) carries determinants for attenuation and virulence in mice. J. Virol. 2000, 74, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Struthers, J.K.; Swanepoel, R. Identification of a major non-structural protein in the nuclei of Rift Valley fever virus-infected cells. J. Gen. Virol. 1982, 60, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Yadani, F.Z.; Kohl, A.; Prehaud, C.; Billecocq, A.; Bouloy, M. The carboxy-terminal acidic domain of Rift Valley fever virus NSs protein is essential for the formation of filamentous structures but not for the nuclear localization of the protein. J. Virol. 1999, 73, 5018–5025. [Google Scholar] [PubMed]
- Muller, R.; Saluzzo, J.F.; Lopez, N.; Dreier, T.; Turell, M.; Smith, J.; Bouloy, M. Characterization of Clone-13, a naturally attenuated avirulent isolate of Rift Valley fever virus, which is altered in the small segment. Am. J. Trop. Med. Hyg. 1995, 53, 405–411. [Google Scholar] [PubMed]
- Billecocq, A.; Spiegel, M.; Vialat, P.; Kohl, A.; Weber, F.; Bouloy, M.; Haller, O. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J. Virol. 2004, 78, 9798–9806. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Dubaele, S.; Proietti De Santis, L.; Billecocq, A.; Bouloy, M.; Egly, J.M. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell 2004, 116, 541–550. [Google Scholar] [CrossRef]
- Kalveram, B.; Lihoradova, O.; Ikegami, T. NSs protein of Rift Valley fever virus promotes posttranslational downregulation of the TFIIH subunit p62. J. Virol. 2011, 85, 6234–6243. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Kandasamy, K.; Alvisi, G.; Mulhern, O.; Sacco, R.; Habjan, M.; Binder, M.; Stefanovic, A.; Eberle, C.A.; Goncalves, A.; et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 2012, 487, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Hermand, D. F-box proteins: More than baits for the SCF? Cell Div. 2006, 1, 30. [Google Scholar] [CrossRef] [PubMed]
- Kainulainen, M.; Habjan, M.; Hubel, P.; Busch, L.; Lau, S.; Colinge, J.; Superti-Furga, G.; Pichlmair, A.; Weber, F. Virulence factor NSs of Rift Valley fever virus recruits the F-box protein FBXO3 to degrade subunit p62 of general transcription factor TFIIH. J. Virol. 2014, 88, 3464–3473. [Google Scholar] [CrossRef] [PubMed]
- Cyr, N.; de la Fuente, C.; Lecoq, L.; Guendel, I.; Chabot, P.R.; Kehn-Hall, K.; Omichinski, J.G. A Omega XaV motif in the Rift Valley fever virus NSs protein is essential for degrading p62, forming nuclear filaments and virulence. Proc. Natl. Acad. Sci. USA 2015, 112, 6021–6026. [Google Scholar] [CrossRef] [PubMed]
- Copeland, A.M.; van Deusen, N.M.; Schmaljohn, C.S. Rift Valley fever virus NSs gene expression correlates with a defect in nuclear mRNA export. Virology 2015, 486, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Mansuroglu, Z.; Leger, P.; Josse, T.; Blot, G.; Billecocq, A.; Flick, R.; Jacob, Y.; Bonnefoy, E.; Bouloy, M. A SAP30 complex inhibits IFN-beta expression in Rift valley fever virus infected cells. PLoS Pathog. 2008, 4, e13. [Google Scholar] [CrossRef] [PubMed]
- Kainulainen, M.; Lau, S.; Samuel, C.E.; Hornung, V.; Weber, F. NSs virulence factor of Rift Valley fever virus engages the F-box proteins FBXW11 and beta-TRCP1 to degrade the antiviral protein kinase PKR. J. Virol. 2016, 90, 6140–6147. [Google Scholar] [CrossRef] [PubMed]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Kota, K.P.; Whitehouse, C.A.; Bavari, S. Protein kinase R degradation is essential for Rift Valley fever virus infection and is regulated by SKP1-CUL1-F-box (SCF)FBXW11-NSs E3 Ligase. PLoS Pathog. 2016, 12, e1005437. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.W., Jr.; Slayter, M.V.; Hall, W.; Peters, C.J. Pathogenesis of a phleboviral infection (Punta Toro virus) in golden Syrian hamsters. Arch. Virol. 1990, 114, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.A.; Narayanan, K.; Worthy, M.; Peters, C.J. The S segment of Punta Toro virus (Bunyaviridae, Phlebovirus) is a major determinant of lethality in the Syrian hamster and codes for a type I interferon antagonist. J. Virol. 2007, 81, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Lihoradova, O.A.; Indran, S.V.; Kalveram, B.; Lokugamage, N.; Head, J.A.; Gong, B.; Tigabu, B.; Juelich, T.L.; Freiberg, A.N.; Ikegami, T. Characterization of Rift Valley fever virus MP-12 strain encoding NSs of Punta Toro virus or sandfly fever Sicilian virus. PLoS Negl. Trop. Dis. 2013, 7, e2181. [Google Scholar] [CrossRef] [PubMed]
- Kalveram, B.; Ikegami, T. Toscana virus NSs protein promotes degradation of double-stranded RNA-dependent protein kinase. J. Virol. 2013, 87, 3710–3718. [Google Scholar] [CrossRef] [PubMed]
- Di Bonito, P.; Nicoletti, L.; Mochi, S.; Accardi, L.; Marchi, A.; Giorgi, C. Immunological characterization of Toscana virus proteins. Arch. Virol. 1999, 144, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Savellini, G.G.; Weber, F.; Terrosi, C.; Habjan, M.; Martorelli, B.; Cusi, M.G. Toscana virus induces interferon although its NSs protein reveals antagonistic activity. J. Gen. Virol. 2011, 92, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Gori-Savellini, G.; Valentini, M.; Cusi, M.G. Toscana Virus NSs Protein Inhibits the Induction of Type I Interferon by Interacting with RIG-I. J. Virol. 2013, 87, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Savellini, G.G.; Gandolfo, C.; Cusi, M.G. Truncation of the C-terminal region of Toscana Virus NSs protein is critical for interferon-beta antagonism and protein stability. Virology 2015, 486, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Brisbarre, N.M.; Plumet, S.; de Micco, P.; Leparc-Goffart, I.; Emonet, S.F. Toscana virus inhibits the interferon beta response in cell cultures. Virology 2013, 442, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Qi, X.; Wu, X.; Liang, M.; Li, C.; Cardona, C.J.; Xu, W.; Tang, F.; Li, Z.; Wu, B.; et al. Suppression of the interferon and NF-kappaB responses by severe fever with thrombocytopenia syndrome virus. J. Virol. 2012, 86, 8388–8401. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.J.; Wang, M.; Deng, M.; Shen, S.; Liu, W.; Cao, W.C.; Deng, F.; Wang, Y.Y.; Hu, Z.; Wang, H. Viral suppression of innate immunity via spatial isolation of TBK1/IKKepsilon from mitochondrial antiviral platform. J. Mol. Cell Biol. 2014, 6, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Santiago, F.W.; Covaleda, L.M.; Sanchez-Aparicio, M.T.; Silvas, J.A.; Diaz-Vizarreta, A.C.; Patel, J.R.; Popov, V.; Yu, X.J.; Garcia-Sastre, A.; Aguilar, P.V. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J. Virol. 2014, 88, 4572–4585. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.D.; Qi, X.; Qu, B.Q.; Zhang, Z.R.; Liang, M.F.; Li, C.; Cardona, C.J.; Li, D.X.; Xing, Z. Evasion of antiviral immunity through sequestering of TBK1/IKK epsilon/IRF3 into viral inclusion bodies. J. Virol. 2014, 88, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.D.; Qi, X.; Liang, M.F.; Li, C.; Cardona, C.J.; Li, D.X.; Xing, Z. Roles of viroplasm-like structures formed by nonstructural protein NSs in infection with severe fever with thrombocytopenia syndrome virus. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 2504–2516. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.J.; Takeuchi, O.; Akira, S.; Chen, Z.J.; Inoue, S.S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.J.; Feng, K.; Min, Y.Q.; Cao, W.C.; Wang, M.; Deng, F.; Hu, Z.; Wang, H. Disruption of type I interferon signaling by the nonstructural protein of severe fever with thrombocytopenia syndrome virus via the hijacking of STAT2 and STAT1 into inclusion bodies. J. Virol. 2015, 89, 4227–4236. [Google Scholar] [CrossRef] [PubMed]
- Simons, J.F.; Hellman, U.; Pettersson, R.F. Uukuniemi virus S RNA Segment—Ambisense coding strategy, packaging of complementary strands into virions, and homology to members of the genus Phlebovirus. J. Virol. 1990, 64, 247–255. [Google Scholar] [PubMed]
- Rezelj, V.V.; Overby, A.K.; Elliott, R.M. Generation of mutant Uukuniemi viruses lacking the nonstructural protein NSs by reverse genetics indicates that NSs is a weak interferon antagonist. J. Virol. 2015, 89, 4849–4856. [Google Scholar] [CrossRef] [PubMed]
- Brennan, B.; Li, P.; Zhang, S.; Li, A.; Liang, M.; Li, D.; Elliott, R.M. Reverse genetics system for severe fever with thrombocytopenia syndrome virus. J. Virol. 2015, 89, 3026–3037. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Won, S.; Peters, C.J.; Makino, S. Rift Valley fever virus NSs mRNA is transcribed from an incoming anti-viral-sense S RNA segment. J. Virol. 2005, 79, 12106–12111. [Google Scholar] [CrossRef] [PubMed]
- Billecocq, A.; Gauliard, N.; le May, N.; Elliott, R.M.; Flick, R.; Bouloy, M. RNA polymerase I-mediated expression of viral RNA for the rescue of infectious virulent and avirulent Rift Valley fever viruses. Virology 2008, 378, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Penski, N.; Spiegel, M.; Weber, F. T7 RNA polymerase-dependent and -independent systems for cDNA-based rescue of Rift Valley fever virus. J. Gen. Virol. 2008, 89, 2157–2166. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Won, S.; Peters, C.J.; Makino, S. Rescue of infectious Rift Valley fever virus entirely from cDNA, analysis of virus lacking the NSs gene, and expression of a foreign gene. J. Virol. 2006, 80, 2933–2940. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ISG | Affected Step in Replication | Affected Phleboviruses (Strains) | References |
|---|---|---|---|
| IFITM2, 3 | uncoating | RVFV (ZH501, MP12) | [110] |
| Mx | primary and secondary transcription | RVFV (MP12, Clone 13), TOSV, SFSV | [106,107,108] |
| OASL2 | ? | RVFV (rZH548ΔNSs) | [100] |
| PKR | viral protein translation | NSs-deficient RVFV mutants (e.g., Clone 13) | [93,111] |
| IFIT1-3 | viral protein translation | RVFV (Clone 13) | [113] |
| mPARP12 | ? | RVFV (MP12) | [112] |
| ISG15 | ? | RVFV (rZH548ΔNSs) | [100] |
| Phlebovirus | Host Target | Mechanism | References |
|---|---|---|---|
| RVFV | TFIIH p44, XPB | sequestration | [119] |
| TFIIH p62 | proteasomal degradation by recruitment of a SKP1-FBXO3 E3-ubiquitin ligase complex | [120,123,124] | |
| SAP30 | recruitment of suppressors to the IFN promoter | [126] | |
| mRNA export | unknown | [125] | |
| PKR | proteasomal degradation by recruitment of a SKP1-CUL1-FBXW11 E3 ligase complex | [93,111,127,128] | |
| TOSV | RIG-I | proteasomal degradation | [134,135,136] |
| PKR | proteasomal degradation | [132] | |
| PTV | IFN induction | unknown | [131] |
| SFSV | IFN induction | unknown | [93,131] |
| Phlebovirus | Host Target | Mechanism | References |
|---|---|---|---|
| SFTSV | RIG-I, TRIM25, TBK1/IKKε, IRF3 | sequestration into cytoplasmic inclusion bodies | [138,139,140,141] |
| STAT1, STAT2 | sequestration into cytoplasmic inclusion bodies | [144] | |
| UUKV | unknown | unknown | [146] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wuerth, J.D.; Weber, F. Phleboviruses and the Type I Interferon Response. Viruses 2016, 8, 174. https://doi.org/10.3390/v8060174
Wuerth JD, Weber F. Phleboviruses and the Type I Interferon Response. Viruses. 2016; 8(6):174. https://doi.org/10.3390/v8060174
Chicago/Turabian StyleWuerth, Jennifer Deborah, and Friedemann Weber. 2016. "Phleboviruses and the Type I Interferon Response" Viruses 8, no. 6: 174. https://doi.org/10.3390/v8060174
APA StyleWuerth, J. D., & Weber, F. (2016). Phleboviruses and the Type I Interferon Response. Viruses, 8(6), 174. https://doi.org/10.3390/v8060174