
The Role of Phlebovirus Glycoproteins in Viral Entry, Assembly and Release



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of Gn and Gc in Phlebovirus Entry
2.1. Configuration of Gn and Gc Proteins in the Viral Envelope
2.2. Attachment Factors and Receptors
2.3. Dendritic Cell-Specific Intercellular Adhesion Molecule-3-Grabbing Non-Integrin (DC-SIGN) Facilitates Phlebovirus Entry into DCs
2.4. Heparan Sulfate (HS) Proteoglycans Promote Phlebovirus Attachment
2.5. Non-Muscle Myosin Heavy Chain IIA (NMMHC-IIA) Promotes SFTSV Entry
2.6. Phlebovirus Uptake: Clathrin-Dependent and -Independent Mechanisms
2.7. Virus-Cell Fusion and Its Inhibition
2.7.1. Characteristics of Viral Membrane Fusion Proteins
2.7.2. RVFV Gc is a Class II Membrane Fusion Protein
2.7.3. Low pH Triggers Membrane Fusion
2.7.4. Inhibition of Membrane Fusion by Interferon-Induced Transmembrane (IFITM) Proteins
3. Role of Gn and Gc in Phlebovirus Assembly
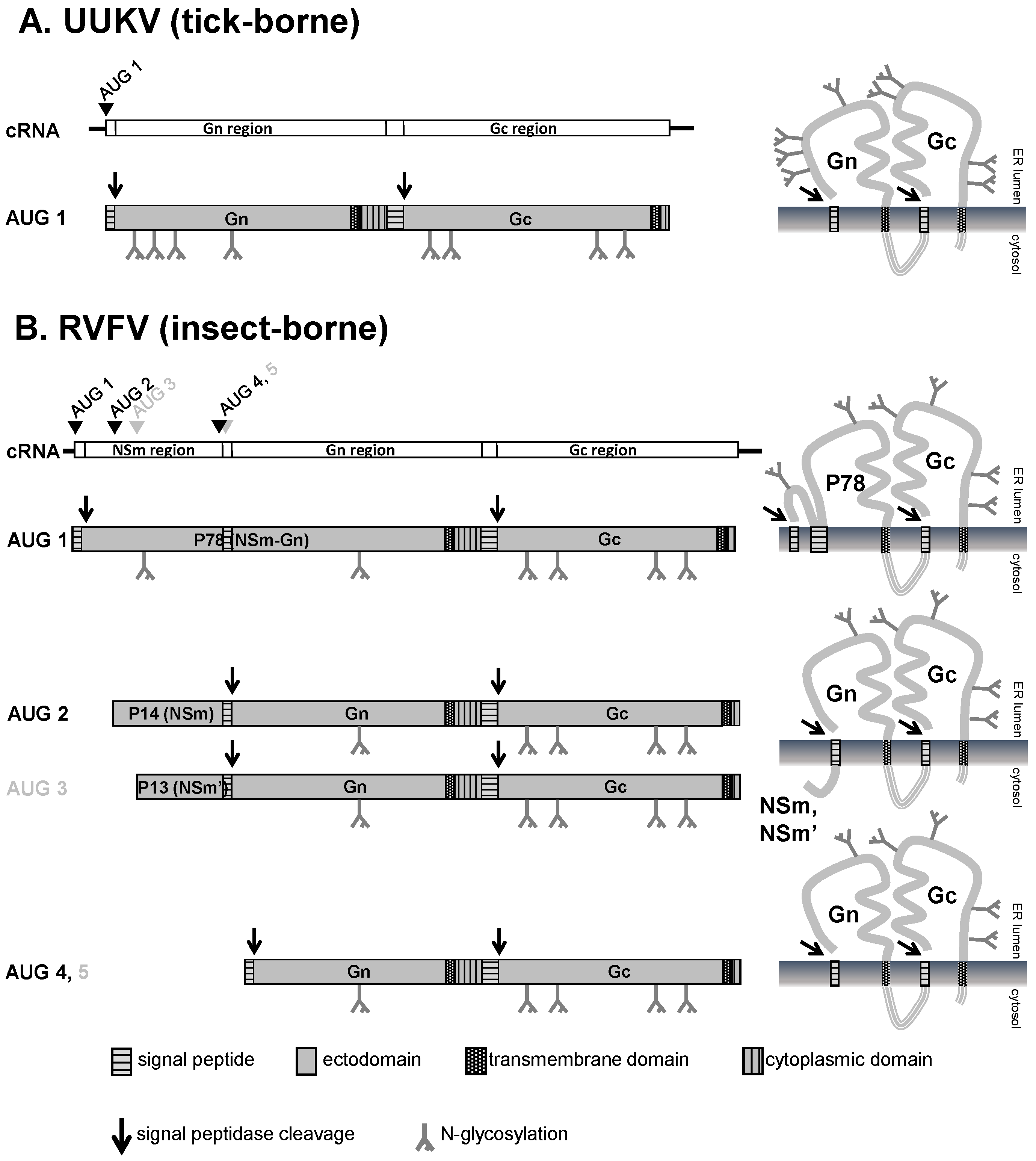
3.1. M Segment Coding Strategy and Expression of the Glycoproteins Gn and Gc
3.2. Post-translational Modifications and Subcellular Localization of Gn and Gc
3.3. The Role of the Cytoplasmic Tails of Gn and Gc in Virus Assembly and Budding
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ANDV | Andes virus |
| BiP | Bindung immunoglobulin protein |
| CavME | Caveolin-1-mediated endocytosis |
| CIE | Clathrin-independent endocytosis |
| CME | Clathrin-mediated endocytosis |
| CNX | Calnexin |
| CCHFV | Crimean Congo hemorrhagic fever virus |
| DC | Dendritic cell |
| DC-SIGN | Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin |
| Env | Envelope glycoprotein |
| ER | Endoplasmic reticulum |
| GAG | Glycosaminoglycan |
| gRNA | genomic RNA |
| HDAC 8 | Histone deacetylase 8 |
| HRTV | Heartland virus |
| HIV | Human immunodeficiency virus |
| HNTV | Hantaan virus |
| HS | Heparan sulfate |
| IFITM | Interferon-induced transmembrane protein |
| L-SIGN | Liver/lymph node-specific intercellular adhesion molecules-3 grabbing non-integrin |
| LACV | La Crosse virus |
| MФ | Macrophage |
| NMMHC-IIA | Non-muscle myosin heavy chain IIA |
| ORF | Open reading frame |
| PDI | Protein disulfide isomerase |
| PTV | Punta Toro virus |
| RNaseK | Ribonuclease kappa |
| RNP | Ribonucleoprotein |
| RVFV | Rift Valley fever virus |
| SFV | Sandfly fever virus |
| SFTSV | Severe fever with thrombocytopenia virus |
| SIV | Simian immunodeficiency virus |
| TOSV | Toscana virus |
| UUKV | Uukuniemi virus |
| v-SNARE | Vesicle-soluble NSF attachment protein receptor |
| VAMP3 | Vesicle-associated membrane protein 3 |
| VSV | Vesicular stomatitis virus |
References
- Plyusnin, A.; Beaty, B.J.; Elliott, R.M.; Goldbach, R.; Kormelink, R.; Lundkvist, Å.; Schmaljohn, C.S.; Tesh, R.B. Bunyaviridae. In Virus Taxonomy. Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Lefkowitz, E., Adams, M.J., Carstens, E.B., Eds.; Academic Press: London/Waltham, UK; San Diego, CA, USA, 2012; pp. 724–741. [Google Scholar]
- Horne, K.M.; Vanlandingham, D.L. Bunyavirus-vector interactions. Viruses 2014, 6, 4373–4397. [Google Scholar] [CrossRef] [PubMed]
- Rotenberg, D.; Jacobson, A.L.; Schneweis, D.J.; Whitfield, A.E. Thrips transmission of tospoviruses. Curr. Opin. Virol. 2015, 15, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Barriere, P.; Koivogui, L.; ter Meulen, J.; Kruger, D.H. Novel Hantavirus sequences in Shrew, Guinea. Emerg. Infect. Dis. 2007, 13, 520–522. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kadjo, B.; Dubey, S.; Jacquet, F.; Yanagihara, R. Molecular evolution of Azagny virus, a newfound Hantavirus harbored by the West African pygmy shrew (Crocidura obscurior) in Cote d’Ivoire. Virol. J. 2011, 8, 373. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Witkowski, P.T.; Auste, B.; Nowak, K.; Weber, N.; Fahr, J.; Mombouli, J.V.; Wolfe, N.D.; Drexler, J.F.; Drosten, C.; et al. Hantavirus in bat, Sierra Leone. Emerg. Infect. Dis. 2012, 18, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Carey, D.E.; Reuben, R.; Panicker, K.N.; Shope, R.E.; Myers, R.M. Thottapalayam virus: A presumptive arbovirus isolated from a shrew in India. Indian J. Med. Res. 1971, 59, 1758–1760. [Google Scholar] [PubMed]
- Guo, W.P.; Lin, X.D.; Wang, W.; Tian, J.H.; Cong, M.L.; Zhang, H.L.; Wang, M.R.; Zhou, R.H.; Wang, J.B.; Li, M.H.; et al. Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents. PLoS Pathog. 2013, 9, e1003159. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Ohdachi, S.D.; Asakawa, M.; Kang, H.J.; Mocz, G.; Arikawa, J.; Okabe, N.; Yanagihara, R. Molecular phylogeny of a newfound Hantavirus in the Japanese shrew mole (Urotrichus talpoides). Proc. Natl. Acad. Sci. USA 2008, 105, 16296–16301. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Bennett, S.N.; Hope, A.G.; Cook, J.A.; Yanagihara, R. Shared ancestry between a newfound mole-borne Hantavirus and hantaviruses harbored by cricetid rodents. J. Virol. 2011, 85, 7496–7503. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Zhang, Y.Z. The evolution and emergence of hantaviruses. Curr. Opin. Virol. 2015, 10, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Walter, C.T.; Barr, J.N. Recent advances in the molecular and cellular biology of bunyaviruses. J. Gen. Virol. 2011, 92 (Pt 11), 2467–2484. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Compans, R.W. Oligomerization, transport, and Golgi retention of Punta Toro virus glycoproteins. J. Virol. 1991, 65, 5902–5909. [Google Scholar] [PubMed]
- Antic, D.; Wright, K.E.; Kang, C.Y. Maturation of Hantaan virus glycoproteins G1 and G2. Virology 1992, 189, 324–328. [Google Scholar] [CrossRef]
- Lober, C.; Anheier, B.; Lindow, S.; Klenk, H.D.; Feldmann, H. The Hantaan virus glycoprotein precursor is cleaved at the conserved pentapeptide WAASA. Virology 2001, 289, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, S.R.; Nichol, S.T. Synthesis, proteolytic processing and complex formation of N-terminally nested precursor proteins of the Rift Valley fever virus glycoproteins. Virology 2007, 357, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.M.; Melin, L.; Persson, R.; Raschperger, E.; Wikstrom, L.; Pettersson, R.F. Processing and membrane topology of the spike proteins G1 and G2 of Uukuniemi virus. J. Virol. 1997, 71, 218–225. [Google Scholar] [PubMed]
- Kuismanen, E. Posttranslational processing of Uukuniemi virus glycoproteins G1 and G2. J. Virol. 1984, 51, 806–812. [Google Scholar] [PubMed]
- Matsuoka, Y.; Ihara, T.; Bishop, D.H.; Compans, R.W. Intracellular accumulation of Punta Toro virus glycoproteins expressed from cloned cDNA. Virology 1988, 167, 251–260. [Google Scholar] [CrossRef]
- Madoff, D.H.; Lenard, J. A membrane glycoprotein that accumulates intracellularly: Cellular processing of the large glycoprotein of LaCrosse virus. Cell 1982, 28, 821–829. [Google Scholar] [CrossRef]
- Shi, X.; Brauburger, K.; Elliott, R.M. Role of N-linked glycans on bunyamwera virus glycoproteins in intracellular trafficking, protein folding, and virus infectivity. J. Virol. 2005, 79, 13725–13734. [Google Scholar] [CrossRef] [PubMed]
- Kuismanen, E.; Hedman, K.; Saraste, J.; Pettersson, R.F. Uukuniemi virus maturation: Accumulation of virus particles and viral antigens in the Golgi complex. Mol. Cell. Biol. 1982, 2, 1444–1458. [Google Scholar] [CrossRef] [PubMed]
- Fontana, J.; Lopez-Montero, N.; Elliott, R.M.; Fernandez, J.J.; Risco, C. The unique architecture of Bunyamwera virus factories around the Golgi complex. Cell. Microbiol. 2008, 10, 2012–2028. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.F.; Pifat, D.Y. Morphogenesis of sandfly viruses (Bunyaviridae family). Virology 1982, 121, 61–81. [Google Scholar] [CrossRef]
- Salanueva, I.J.; Novoa, R.R.; Cabezas, P.; Lopez-Iglesias, C.; Carrascosa, J.L.; Elliott, R.M.; Risco, C. Polymorphism and structural maturation of bunyamwera virus in Golgi and post-Golgi compartments. J. Virol. 2003, 77, 1368–1381. [Google Scholar] [CrossRef] [PubMed]
- Murphy, F.A.; Harrison, A.K.; Whitfield, S.G. Bunyaviridae: Morphologic and morphogenetic similarities of Bunyamwera serologic supergroup viruses and several other arthropod-borne viruses. Intervirology 1973, 1, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Piper, M.E.; Sorenson, D.R.; Gerrard, S.R. Efficient cellular release of Rift Valley fever virus requires genomic RNA. PLoS ONE 2011, 6, e18070. [Google Scholar] [CrossRef] [PubMed]
- Overby, A.K.; Popov, V.; Neve, E.P.; Pettersson, R.F. Generation and analysis of infectious virus-like particles of Uukuniemi virus (Bunyaviridae): A useful system for studying bunyaviral packaging and budding. J. Virol. 2006, 80, 10428–10435. [Google Scholar] [CrossRef] [PubMed]
- Novoa, R.R.; Calderita, G.; Cabezas, P.; Elliott, R.M.; Risco, C. Key Golgi factors for structural and functional maturation of bunyamwera virus. J. Virol. 2005, 79, 10852–10863. [Google Scholar] [CrossRef] [PubMed]
- Spiropoulou, C.F. Hantavirus maturation. In Hantaviruses; Schmaljohn, C.S., Nichol, S.T., Eds.; Springer-Verlag: Heidelberg/Berlin, Germany, 2001; pp. 33–46. [Google Scholar]
- Cifuentes-Muñoz, N.; Salazar-Quiroz, N.; Tischler, N.D. Hantavirus Gn and Gc envelope glycoproteins: Key structural units for virus cell entry and virus assembly. Viruses 2014, 6, 1801–1822. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, G.; Lopez-Gil, E.; Warimwe, G.M.; Brun, A. Understanding Rift Valley fever: Contributions of animal models to disease characterization and control. Mol. Immunol. 2015, 66, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chai, C.; Wang, C.; Amer, S.; Lv, H.; He, H.; Sun, J.; Lin, J. Systematic review of severe fever with thrombocytopenia syndrome: Virology, epidemiology, and clinical characteristics. Rev. Med. Virol. 2014, 24, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Q.; Hu, W.; Wu, J.; Wang, Y.; Mei, L.; Walker, D.H.; Ren, J.; Wang, Y.; Yu, X.J. Person-to-person transmission of severe fever with thrombocytopenia syndrome virus. Vector Borne Zoonotic Dis. 2012, 12, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Bichaud, L.; de Lamballerie, X.; Alten, B.; Gould, E.A.; Charrel, R.N. Sandfly-borne phleboviruses of Eurasia and Africa: Epidemiology, genetic diversity, geographic range, control measures. Antivir. Res. 2013, 100, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M.; Brennan, B. Emerging phleboviruses. Curr. Opin. Virol. 2014, 5, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.L.; Lindsey-Regnery, H.; Sasso, D.R.; McCormick, J.B.; Palmer, E. Distinction between Bunyaviridae genera by surface structure and comparison with Hantaan virus using negative stain electron microscopy. Arch. Virol. 1985, 86, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Overby, A.K.; Pettersson, R.F.; Grunewald, K.; Huiskonen, J.T. Insights into bunyavirus architecture from electron cryotomography of Uukuniemi virus. Proc. Natl. Acad. Sci. USA 2008, 105, 2375–2379. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, A.N.; Sherman, M.B.; Morais, M.C.; Holbrook, M.R.; Watowich, S.J. Three-dimensional organization of Rift Valley fever virus revealed by cryoelectron tomography. J. Virol. 2008, 82, 10341–10348. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.B.; Freiberg, A.N.; Holbrook, M.R.; Watowich, S.J. Single-particle cryo-electron microscopy of Rift Valley fever virus. Virology 2009, 387, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Huiskonen, J.T.; Overby, A.K.; Weber, F.; Grunewald, K. Electron cryo-microscopy and single-particle averaging of Rift Valley fever virus: Evidence for GN-GC glycoprotein heterodimers. J. Virol. 2009, 83, 3762–3799. [Google Scholar] [CrossRef] [PubMed]
- Rusu, M.; Bonneau, R.; Holbrook, M.R.; Watowich, S.J.; Birmanns, S.; Wriggers, W.; Freiberg, A.N. An assembly model of Rift Valley fever virus. Front. Microbiol. 2012, 3, 254. [Google Scholar] [CrossRef] [PubMed]
- Dessau, M.; Modis, Y. Crystal structure of glycoprotein C from Rift Valley fever virus. Proc. Natl. Acad. Sci. USA 2013, 110, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Raftery, M.J.; Lalwani, P.; Krautkrmer, E.; Peters, T.; Scharffetter-Kochanek, K.; Kruger, R.; Hofmann, J.; Seeger, K.; Kruger, D.H.; Schonrich, G. Beta2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J. Exp. Med. 2014, 211, 1485–1497. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.N.; Shepley, M.; Shaw, R.; Ginsberg, M.H.; Mackow, E.R. Beta3 integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl. Acad. Sci. USA 1998, 95, 7074–7079. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.N.; Brown, E.J.; Ginsberg, M.H.; Mackow, E.R. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J. Virol. 1999, 73, 3951–3959. [Google Scholar] [PubMed]
- Garcia-Vallejo, J.J.; van Kooyk, Y. The physiological role of DC-SIGN: A tale of mice and men. Trends Immunol. 2013, 34, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, H.; Mitchell, D.A.; Drickamer, K.; Weis, W.I. Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science 2001, 294, 2163–2166. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; van Vliet, S.J.; Koppel, E.A.; Sanchez-Hernandez, M.; Vandenbroucke-Grauls, C.M.; Appelmelk, B.; van Kooyk, Y. Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 2003, 197, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tailleux, L.; Schwartz, O.; Herrmann, J.L.; Pivert, E.; Jackson, M.; Amara, A.; Legres, L.; Dreher, D.; Nicod, L.P.; Gluckman, J.C.; et al. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 2003, 197, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Lozach, P.Y.; Kuhbacher, A.; Meier, R.; Mancini, R.; Bitto, D.; Bouloy, M.; Helenius, A. DC-SIGN as a receptor for phleboviruses. Cell Host Microbe 2011, 10, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Li, X.; Zhang, X.; Liu, W.; Kuhl, A.; Kaup, F.; Soldan, S.S.; Gonzalez-Scarano, F.; Weber, F.; He, Y.; et al. Severe fever with thrombocytopenia virus glycoproteins are targeted by neutralizing antibodies and can use DC-SIGN as a receptor for pH-dependent entry into human and animal cell lines. J. Virol. 2013, 87, 4384–4394. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Sanchez, E.; Altmeyer, R.; Amara, A.; Schwartz, O.; Fieschi, F.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Despres, P. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003, 4, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Tassaneetrithep, B.; Burgess, T.H.; Granelli-Piperno, A.; Trumpfheller, C.; Finke, J.; Sun, W.; Eller, M.A.; Pattanapanyasat, K.; Sarasombath, S.; Birx, D.L.; et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003, 197, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Cerny, D.; Haniffa, M.; Shin, A.; Bigliardi, P.; Tan, B.K.; Lee, B.; Poidinger, M.; Tan, E.Y.; Ginhoux, F.; Fink, K. Selective susceptibility of human skin antigen presenting cells to productive dengue virus infection. PLoS Pathog. 2014, 10, e1004548. [Google Scholar] [CrossRef] [PubMed]
- Engering, A.; Geijtenbeek, T.B.; van Vliet, S.J.; Wijers, M.; Van, L.E.; Demaurex, N.; Lanzavecchia, A.; Fransen, J.; Figdor, C.G.; Piguet, V.; et al. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J. Immunol. 2002, 168, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Bashirova, A.A.; Geijtenbeek, T.B.; van Duijnhoven, G.C.; van Vliet, S.J.; Eilering, J.B.; Martin, M.P.; Wu, L.; Martin, T.D.; Viebig, N.; Knolle, P.A.; et al. A dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN)-related protein is highly expressed on human liver sinusoidal endothelial cells and promotes HIV-1 infection. J. Exp. Med. 2001, 193, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Gramberg, T.; Hofmann, H.; Moller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005, 340, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, S.; Soilleux, E.J.; Baribaud, F.; Leslie, G.J.; Morris, L.S.; Trowsdale, J.; Lee, B.; Coleman, N.; Doms, R.W. DC-SIGNR, a DC-SIGN homologue expressed in endothelial cells, binds to human and simian immunodeficiency viruses and activates infection in trans. Proc. Natl. Acad. Sci. USA 2001, 98, 2670–2675. [Google Scholar] [CrossRef] [PubMed]
- Leger, P.; Tetard, M.; Youness, B.; Cordes, N.; Rouxel, R.N.; Flamand, M.; Lozach, P.Y. Differential use of the C-type lectins L-SIGN and DC-SIGN for phlebovirus endocytosis. Traffic 2016, 17, 639–656. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Feinberg, H.; Conroy, E.; Mitchell, D.A.; Alvarez, R.; Blixt, O.; Taylor, M.E.; Weis, W.I.; Drickamer, K. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat. Struct. Mol. Biol. 2004, 11, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Liang, M.; Ning, J.; Gu, W.; Jiang, H.; Wu, W.; Zhang, F.; Li, C.; Zhang, Q.; Zhu, H.; et al. Pathogenesis of emerging severe fever with thrombocytopenia syndrome virus in C57/BL6 mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 10053–10058. [Google Scholar] [CrossRef] [PubMed]
- Chaipan, C.; Soilleux, E.J.; Simpson, P.; Hofmann, H.; Gramberg, T.; Marzi, A.; Geier, M.; Stewart, E.A.; Eisemann, J.; Steinkasserer, A.; et al. DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J. Virol. 2006, 80, 8951–8960. [Google Scholar] [CrossRef] [PubMed]
- De Boer, S.M.; Kortekaas, J.; de Haan, C.A.; Rottier, P.J.; Moormann, R.J.; Bosch, B.J. Heparan sulfate facilitates Rift Valley fever virus entry into the cell. J. Virol. 2012, 86, 13767–13771. [Google Scholar] [CrossRef] [PubMed]
- Weingartl, H.M.; Zhang, S.; Marszal, P.; McGreevy, A.; Burton, L.; Wilson, W.C. Rift Valley fever virus incorporates the 78 kDa glycoprotein into virions matured in mosquito C6/36 cells. PLoS ONE 2014, 9, e87385. [Google Scholar] [CrossRef] [PubMed]
- Kreher, F.; Tamietti, C.; Gommet, C.; Guillemot, L.; Ermonval, M.; Failloux, A.B.; Panthier, J.J.; Bouloy, M.; Flamand, M. The Rift Valley fever accessory proteins NSm and P78/NSm-GN are distinct determinants of virus propagation in vertebrate and invertebrate hosts. Emerg. Microbes Infect. 2014, 3, e71. [Google Scholar] [CrossRef] [PubMed]
- Riblett, A.M.; Blomen, V.A.; Jae, L.T.; Altamura, L.A.; Doms, R.W.; Brummelkamp, T.R.; Wojcechowskyj, J.A. A haploid genetic screen identifies heparan sulfate proteoglycans supporting Rift Valley fever virus infection. J. Virol. 2015, 90, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Pietrantoni, A.; Fortuna, C.; Remoli, M.E.; Ciufolini, M.G.; Superti, F. Bovine lactoferrin inhibits Toscana virus infection by binding to heparan sulphate. Viruses 2015, 7, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Qi, Y.; Liu, C.; Gao, W.; Chen, P.; Fu, L.; Peng, B.; Wang, H.; Jing, Z.; Zhong, G.; et al. Nonmuscle myosin heavy chain IIA is a critical factor contributing to the efficiency of early infection of severe fever with thrombocytopenia syndrome virus. J. Virol. 2014, 88, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Althaus, K.; Greinacher, A. MYH9-related platelet disorders. Semin. Thromb. Hemost. 2009, 35, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Arii, J.; Goto, H.; Suenaga, T.; Oyama, M.; Kozuka-Hata, H.; Imai, T.; Minowa, A.; Akashi, H.; Arase, H.; Kawaoka, Y.; et al. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 2010, 467, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Lozach, P.Y.; Mancini, R.; Bitto, D.; Meier, R.; Oestereich, L.; Overby, A.K.; Pettersson, R.F.; Helenius, A. Entry of bunyaviruses into mammalian cells. Cell Host Microbe 2010, 7, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Franceschini, A.; Horvath, P.; Tetard, M.; Mancini, R.; von Mering, C.; Helenius, A.; Lozach, P.Y. Genome-wide small interfering RNA screens reveal VAMP3 as a novel host factor required for Uukuniemi virus late penetration. J. Virol. 2014, 88, 8565–8578. [Google Scholar] [CrossRef] [PubMed]
- Proux-Gillardeaux, V.; Rudge, R.; Galli, T. The tetanus neurotoxin-sensitive and insensitive routes to and from the plasma membrane: Fast and slow pathways? Traffic 2005, 6, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Sanchez, D.G.; Mestre, M.B.; Colombo, M.I. TI-VAMP/VAMP7 and VAMP3/cellubrevin: Two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim. Biophys. Acta 2009, 1793, 1901–1916. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Boukari, H.; Banerjee, I.; Sbalzarini, I.F.; Horvath, P.; Helenius, A. Histone deacetylase 8 is required for centrosome cohesion and influenza A virus entry. PLoS Pathog. 2011, 7, e1002316. [Google Scholar] [CrossRef] [PubMed]
- Hackett, B.A.; Yasunaga, A.; Panda, D.; Tartell, M.A.; Hopkins, K.C.; Hensley, S.E.; Cherry, S. RNASEK is required for internalization of diverse acid-dependent viruses. Proc. Natl. Acad. Sci. USA 2015, 112, 7797–7802. [Google Scholar] [CrossRef] [PubMed]
- Harmon, B.; Schudel, B.R.; Maar, D.; Kozina, C.; Ikegami, T.; Tseng, C.T.; Negrete, O.A. Rift Valley fever virus strain MP-12 enters mammalian host cells via caveola-mediated endocytosis. J. Virol. 2012, 86, 12954–12970. [Google Scholar] [CrossRef] [PubMed]
- Hollidge, B.S.; Gonzalez-Scarano, F.; Soldan, S.S. Arboviral encephalitides: Transmission, emergence, and pathogenesis. J. Neuroimmune Pharmacol. 2010, 5, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.I.; Rodrigues, A.H.; Silva, M.L.; Mortara, R.A.; Rossi, M.A.; Jamur, M.C.; Oliver, C.; Arruda, E. Oropouche virus entry into HeLa cells involves clathrin and requires endosomal acidification. Virus Res. 2008, 138, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Podbilewicz, B. Virus and cell fusion mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 111–139. [Google Scholar] [CrossRef] [PubMed]
- Vaney, M.C.; Rey, F.A. Class II enveloped viruses. Cell. Microbiol. 2011, 13, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Garry, C.E.; Garry, R.F. Proteomics computational analyses suggest that the carboxyl terminal glycoproteins of Bunyaviruses are class II viral fusion protein (beta-penetrenes). Theor. Biol. Med. Model. 2004, 1, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boer, S.M.; Kortekaas, J.; Spel, L.; Rottier, P.J.; Moormann, R.J.; Bosch, B.J. Acid-activated structural reorganization of the Rift Valley fever virus Gc fusion protein. J. Virol. 2012, 86, 13642–13652. [Google Scholar] [CrossRef] [PubMed]
- Filone, C.M.; Heise, M.; Doms, R.W.; Bertolotti-Ciarlet, A. Development and characterization of a Rift Valley fever virus cell-cell fusion assay using alphavirus replicon vectors. Virology 2006, 356, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Bitto, D.; Halldorsson, S.; Caputo, A.; Huiskonen, J.T. Low pH and anionic lipid dependent fusion of Uukuniemi phlebovirus to liposomes. J. Biol. Chem 2016, 291, 6412–6422. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Weston, S.; Kellam, P.; Marsh, M. IFITM proteins-cellular inhibitors of viral entry. Curr. Opin. Virol. 2014, 4, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. IFITMs restrict the replication of multiple pathogenic viruses. J. Mol. Biol. 2013, 425, 4937–4955. [Google Scholar] [CrossRef] [PubMed]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Radoshitzky, S.R.; Kota, K.P.; Altamura, L.A.; Smith, J.M.; Packard, B.Z.; Kuhn, J.H.; Costantino, J.; et al. IFITM-2 and IFITM-3 but not IFITM-1 restrict Rift Valley fever virus. J. Virol. 2013, 87, 8451–8464. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Markosyan, R.M.; Zheng, Y.M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Mercer, J. Lipid interactions during virus entry and infection. Cell. Microbiol. 2014, 16, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Won, S.; Peters, C.J.; Makino, S. Rift Valley fever virus NSs mRNA is transcribed from an incoming anti-viral-sense S RNA segment. J. Virol. 2005, 79, 12106–12111. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.N. Bunyavirus mRNA synthesis is coupled to translation to prevent premature transcription termination. RNA 2007, 13, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Collett, M.S.; Purchio, A.F.; Keegan, K.; Frazier, S.; Hays, W.; Anderson, D.K.; Parker, M.D.; Schmaljohn, C.; Schmidt, J.; Dalrymple, J.M. Complete nucleotide sequence of the M RNA segment of Rift Valley fever virus. Virology 1985, 144, 228–245. [Google Scholar] [CrossRef]
- Ihara, T.; Smith, J.; Dalrymple, J.M.; Bishop, D.H. Complete sequences of the glycoproteins and M RNA of Punta Toro phlebovirus compared to those of Rift Valley fever virus. Virology 1985, 144, 246–259. [Google Scholar] [CrossRef]
- Ronnholm, R.; Pettersson, R.F. Complete nucleotide sequence of the M RNA segment of Uukuniemi virus encoding the membrane glycoproteins G1 and G2. Virology 1987, 160, 191–202. [Google Scholar] [CrossRef]
- Gro, M.C.; di Bonito, P.; Fortini, D.; Mochi, S.; Giorgi, C. Completion of molecular characterization of Toscana phlebovirus genome: Nucleotide sequence, coding strategy of M genomic segment and its amino acid sequence comparison to other phleboviruses. Virus Res. 1997, 51, 81–91. [Google Scholar] [CrossRef]
- Kuismanen, E.; Bang, B.; Hurme, M.; Pettersson, R.F. Uukuniemi virus maturation: Immunofluorescence microscopy with monoclonal glycoprotein-specific antibodies. J. Virol. 1984, 51, 137–146. [Google Scholar] [PubMed]
- Kakach, L.T.; Wasmoen, T.L.; Collett, M.S. Rift Valley fever virus M segment: Use of recombinant vaccinia viruses to study Phlebovirus gene expression. J. Virol. 1988, 62, 826–833. [Google Scholar] [PubMed]
- Wasmoen, T.L.; Kakach, L.T.; Collett, M.S. Rift Valley fever virus M segment: Cellular localization of M segment-encoded proteins. Virology 1988, 166, 275–280. [Google Scholar] [CrossRef]
- Ulmanen, I.; Seppala, P.; Pettersson, R.F. In vitro translation of Uukuniemi virus-specific RNAs: Identification of a nonstructural protein and a precursor to the membrane glycoproteins. J. Virol. 1981, 37, 72–79. [Google Scholar] [PubMed]
- Suzich, J.A.; Collett, M.S. Rift Valley fever virus M segment: Cell-free transcription and translation of virus-complementary RNA. Virology 1988, 164, 478–486. [Google Scholar] [CrossRef]
- Pettersson, R.F.; Melin, L. Synthesis, assembly, and intracellular transport of Bunyaviridae membrane proteins. In The Bunyaviridae; Elliott, R., Ed.; Plenum Press: New York, NY, USA, 1996; pp. 159–183. [Google Scholar]
- Chen, S.Y.; Matsuoka, Y.; Compans, R.W. Golgi complex localization of the Punta Toro virus G2 protein requires its association with the G1 protein. Virology 1991, 183, 351–365. [Google Scholar] [CrossRef]
- Xu, B.; Liu, L.; Huang, X.; Ma, H.; Zhang, Y.; Du, Y.; Wang, P.; Tang, X.; Wang, H.; Kang, K.; et al. Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan province, China: Discovery of a new bunyavirus. PLoS Pathog. 2011, 7, e1002369. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.J.; Liang, M.F.; Zhang, S.Y.; Liu, Y.; Li, J.D.; Sun, Y.L.; Zhang, L.; Zhang, Q.F.; Popov, V.L.; Li, C.; et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 2011, 364, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Di Bonito, P.; Mochi, S.; Gro, M.C.; Fortini, D.; Giorgi, C. Organization of the M genomic segment of Toscana phlebovirus. J. Gen. Virol. 1997, 78 (Pt 1), 77–81. [Google Scholar] [CrossRef] [PubMed]
- Suzich, J.A.; Kakach, L.T.; Collett, M.S. Expression strategy of a phlebovirus: Biogenesis of proteins from the Rift Valley fever virus M segment. J. Virol. 1990, 64, 1549–1555. [Google Scholar] [PubMed]
- Ikegami, T. Molecular biology and genetic diversity of Rift Valley fever virus. Antivir. Res. 2012, 95, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Ikegami, T.; Peters, C.J.; Makino, S. NSm protein of Rift Valley fever virus suppresses virus-induced apoptosis. J. Virol. 2007, 81, 13335–13345. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Ikegami, T.; Peters, C.J.; Makino, S. NSm and 78-kilodalton proteins of Rift Valley fever virus are nonessential for viral replication in cell culture. J. Virol. 2006, 80, 8274–8278. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, S.R.; Bird, B.H.; Albarino, C.G.; Nichol, S.T. The NSm proteins of Rift Valley fever virus are dispensable for maturation, replication and infection. Virology 2007, 359, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.M.; Pettersson, R.F. Targeting of a short peptide derived from the cytoplasmic tail of the G1 membrane glycoprotein of Uukuniemi virus (Bunyaviridae) to the Golgi complex. J. Virol. 1998, 72, 9585–9596. [Google Scholar] [PubMed]
- Persson, R.; Pettersson, R.F. Formation and intracellular transport of a heterodimeric viral spike protein complex. J. Cell Biol. 1991, 112, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Veijola, J.; Pettersson, R.F. Transient association of calnexin and calreticulin with newly synthesized G1 and G2 glycoproteins of Uukuniemi virus (family Bunyaviridae). J. Virol. 1999, 73, 6123–6127. [Google Scholar] [PubMed]
- Pesonen, M.; Kuismanen, E.; Pettersson, R.F. Monosaccharide sequence of protein-bound glycans of Uukuniemi virus. J. Virol. 1982, 41, 390–400. [Google Scholar] [PubMed]
- Chen, S.Y.; Matsuoka, Y.; Compans, R.W. Assembly of G1 and G2 glycoprotein oligomers in Punta Toro virus-infected cells. Virus Res. 1992, 22, 215–225. [Google Scholar] [PubMed]
- Gahmberg, N.; Kuismanen, E.; Keranen, S.; Pettersson, R.F. Uukuniemi virus glycoproteins accumulate in and cause morphological changes of the Golgi complex in the absence of virus maturation. J. Virol. 1986, 57, 899–906. [Google Scholar] [PubMed]
- Andersson, A.M.; Melin, L.; Bean, A.; Pettersson, R.F. A retention signal necessary and sufficient for Golgi localization maps to the cytoplasmic tail of a Bunyaviridae (Uukuniemi virus) membrane glycoprotein. J. Virol. 1997, 71, 4717–4727. [Google Scholar] [PubMed]
- Gerrard, S.R.; Nichol, S.T. Characterization of the Golgi retention motif of Rift Valley fever virus G(N) glycoprotein. J. Virol. 2002, 76, 12200–12210. [Google Scholar] [CrossRef] [PubMed]
- Ronnholm, R. Localization to the Golgi complex of Uukuniemi virus glycoproteins G1 and G2 expressed from cloned cDNAs. J. Virol. 1992, 66, 4525–4531. [Google Scholar] [PubMed]
- Carnec, X.; Ermonval, M.; Kreher, F.; Flamand, M.; Bouloy, M. Role of the cytosolic tails of Rift Valley fever virus envelope glycoproteins in viral morphogenesis. Virology 2014, 448, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jäntti, J.; Hildén, P.; Rönkä, H.; Mäkiranta, V.; Keränen, S.; Kuismanen, E. Immunocytochemical analysis of Uukuniemi virus budding compartments: Role of the intermediate compartment and the Golgi stack in virus maturation. J. Virol. 1997, 71, 1162–1172. [Google Scholar] [PubMed]
- Overby, A.K.; Popov, V.L.; Pettersson, R.F.; Neve, E.P. The cytoplasmic tails of Uukuniemi Virus (Bunyaviridae) G(N) and G(C) glycoproteins are important for intracellular targeting and the budding of virus-like particles. J. Virol. 2007, 81, 11381–11391. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Chen, S.Y.; Compans, R.W. A signal for Golgi retention in the bunyavirus G1 glycoprotein. J. Biol. Chem. 1994, 269, 22565–22573. [Google Scholar] [PubMed]
- Nilsson, T.; Jackson, M.; Peterson, P.A. Short cytoplasmic sequences serve as retention signals for transmembrane proteins in the endoplasmic reticulum. Cell 1989, 58, 707–718. [Google Scholar] [CrossRef]
- Jackson, M.R.; Nilsson, T.; Peterson, P.A. Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO J. 1990, 9, 3153–3162. [Google Scholar] [PubMed]
- Vincent, M.J.; Martin, A.S.; Compans, R.W. Function of the KKXX motif in endoplasmic reticulum retrieval of a transmembrane protein depends on the length and structure of the cytoplasmic domain. J. Biol. Chem. 1998, 273, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Kakach, L.T.; Suzich, J.A.; Collett, M.S. Rift Valley fever virus M segment: Phlebovirus expression strategy and protein glycosylation. Virology 1989, 170, 505–510. [Google Scholar] [CrossRef]
- Chen, S.Y.; Matsuoka, Y.; Compans, R.W. Assembly and polarized release of Punta Toro virus and effects of brefeldin A. J. Virol. 1991, 65, 1427–1439. [Google Scholar] [PubMed]
- Crispin, M.; Harvey, D.J.; Bitto, D.; Halldorsson, S.; Bonomelli, C.; Edgeworth, M.; Scrivens, J.H.; Huiskonen, J.T.; Bowden, T.A. Uukuniemi Phlebovirus assembly and secretion leave a functional imprint on the virion glycome. J. Virol. 2014, 88, 10244–10251. [Google Scholar] [CrossRef] [PubMed]
- Overby, A.K.; Pettersson, R.F.; Neve, E.P. The glycoprotein cytoplasmic tail of Uukuniemi virus (Bunyaviridae) interacts with ribonucleoproteins and is critical for genome packaging. J. Virol. 2007, 81, 3198–3205. [Google Scholar] [CrossRef] [PubMed]
- Strandin, T.; Hepojoki, J.; Vaheri, A. Cytoplasmic tails of bunyavirus Gn glycoproteins-Could they act as matrix protein surrogates? Virology 2013, 437, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M.; Schmaljohn, C.S. Bunyaviridae. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lipincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1244–1282. [Google Scholar]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiegel, M.; Plegge, T.; Pöhlmann, S. The Role of Phlebovirus Glycoproteins in Viral Entry, Assembly and Release. Viruses 2016, 8, 202. https://doi.org/10.3390/v8070202
Spiegel M, Plegge T, Pöhlmann S. The Role of Phlebovirus Glycoproteins in Viral Entry, Assembly and Release. Viruses. 2016; 8(7):202. https://doi.org/10.3390/v8070202
Chicago/Turabian StyleSpiegel, Martin, Teresa Plegge, and Stefan Pöhlmann. 2016. "The Role of Phlebovirus Glycoproteins in Viral Entry, Assembly and Release" Viruses 8, no. 7: 202. https://doi.org/10.3390/v8070202
APA StyleSpiegel, M., Plegge, T., & Pöhlmann, S. (2016). The Role of Phlebovirus Glycoproteins in Viral Entry, Assembly and Release. Viruses, 8(7), 202. https://doi.org/10.3390/v8070202