Diversity of dsDNA Viruses in a South African Hot Spring Assessed by Metagenomics and Microscopy


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Virus Enrichment
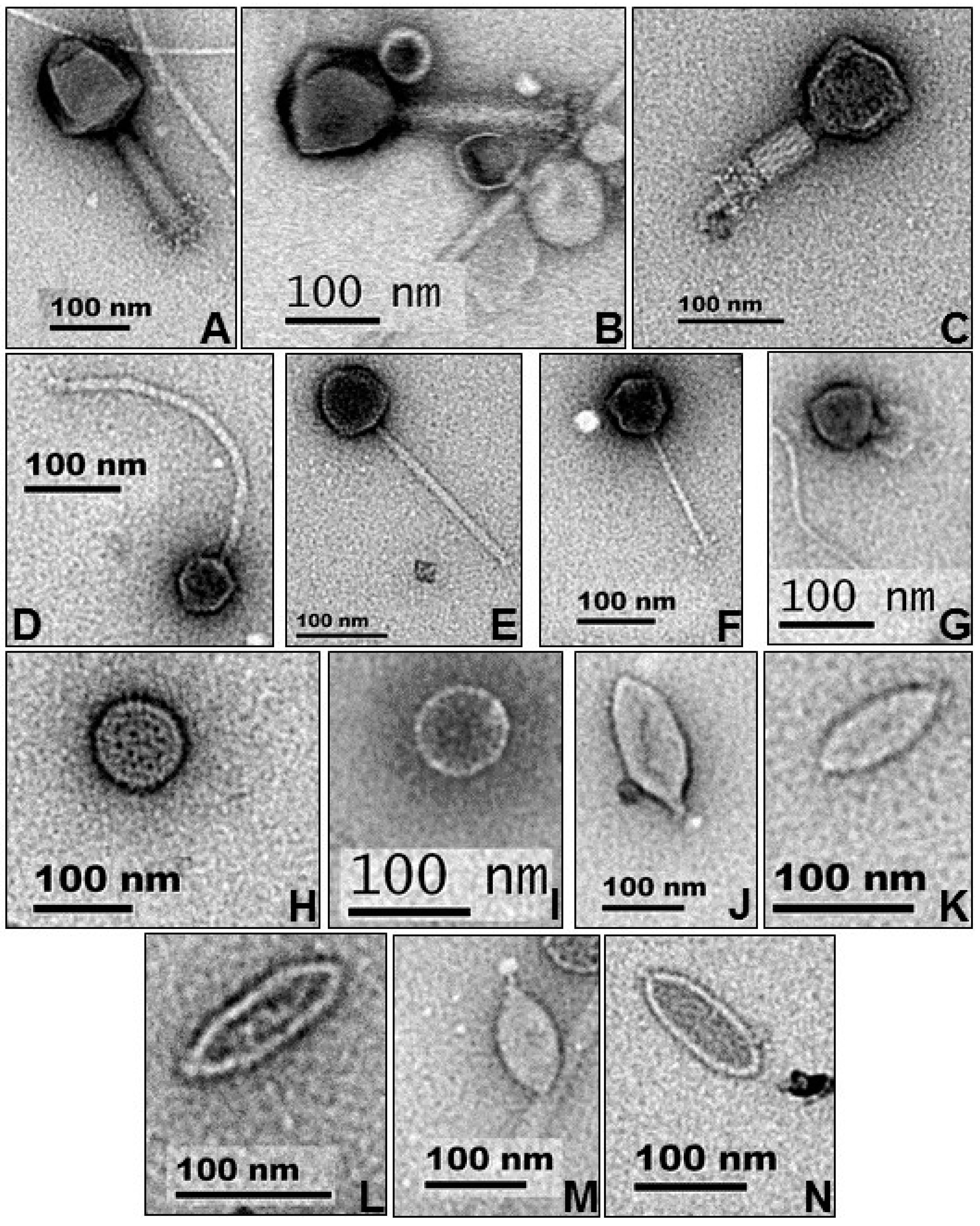
2.2. Electron Microscopy
2.3. Virome DNA Purification, Sequencing and Read Assembly
2.4. Viral Diversity Predictions
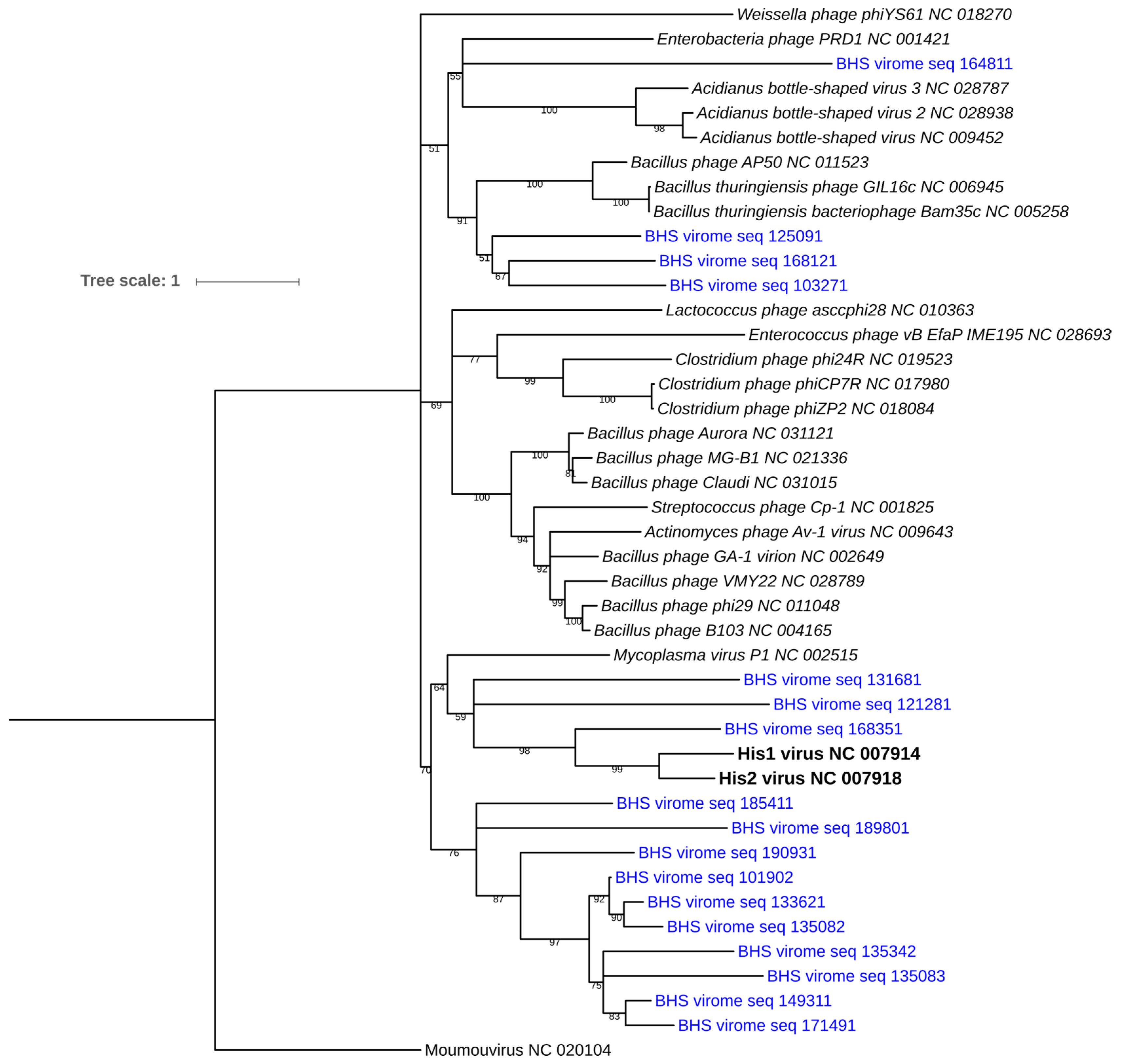
2.5. Phylogenetic Analyses of terL and polB2 Genes
2.6. CRISPR Predictions and Host Assignments
2.7. PCR Amplification, Cloning and Sequencing of the Gemmata-Related Terminase Gene
3. Results
3.1. General Characteristics of Brandvlei Hot Spring
3.2. Electron Microscopy
3.3. Analysis of the Most Represented Viral Genome Fragments
3.4. Diversity of Bacteriophages
3.5. Diversity of Archaeal Viruses
3.5.1. Identification Using the polB2 Gene
3.5.2. Identification of Archaeal Viruses Using Reference Genome Read Mapping
3.5.3. Identification of Archaeal Virus Genes by Using Host Functional Gene Annotations
3.6. CRISPR-Guided Virus-Host Associations
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Inskeep, W.P.; Rusch, D.B.; Jay, Z.J.; Herrgard, M.J.; Kozubal, M.A.; Richardson, T.H.; Macur, R.E.; Hamamura, N.; deM Jennings, R.; Fouke, B.W.; et al. Metagenomes from high-temperature chemotrophic systems reveal geochemical controls on microbial community structure and function. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Shaughnessy, D.P.; Wolf, Y.I.; Koonin, E.V.; Roberto, F.F.; Young, M. Identification of novel positive-strand RNA viruses by metagenomic analysis of archaea-dominated Yellowstone hot springs. J. Virol. 2012, 86, 5562–5573. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, T.; Patterson, M.; Richardson, P.M.; Wommack, K.E.; Young, M.; Mead, D. Assembly of viral metagenomes from Yellowstone hot springs. Appl. Environ. Microbiol. 2008, 74, 4164–4174. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.; Stedman, K.; Snyder, J.; Wiedenheft, B.; Willits, D.; Brumfield, S.; McDermott, T.; Young, M.J. Viruses from extreme thermal environments. Proc. Natl. Acad. Sci. USA 2001, 98, 13341–13345. [Google Scholar] [CrossRef] [PubMed]
- Rachel, R.; Bettstetter, M.; Hedlund, B.P.; Häring, M.; Kessler, A.; Stetter, K.O.; Prangishvili, D. Remarkable morphological diversity of viruses and virus-like particles in hot terrestrial environments. Arch. Virol. 2002, 147, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Gudbergsdóttir, S.R.; Menzel, P.; Krogh, A.; Young, M.; Peng, X. Novel viral genomes identified from six metagenomes reveal wide distribution of archaeal viruses and high viral diversity in terrestrial hot springs. Environ. Microbiol. 2016, 18, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Strazzulli, A.; Fusco, S.; Cobucci-Ponzano, B.; Moracci, M.; Contursi, P. Metagenomics of microbial and viral life in terrestrial geothermal environments. Rev. Environ. Sci. Biotechnol. 2017, 16, 425–454. [Google Scholar] [CrossRef]
- Olivier, J.; Venter, J.S.; Jonker, C.Z. Thermal and chemical characteristics of hot water springs in the northern part of the Limpopo Province, South Africa. Water SA 2011, 37, 427–436. [Google Scholar] [CrossRef]
- Tekere, M.; Prinsloo, A.; Olivier, J.; Jonker, N.; Venter, S. An evaluation of the bacterial diversity at Tshipise, Mphephu and Sagole hot water springs, Limpopo Province, South Africa. Afr. J. Microbiol. Res. 2012, 6, 4993–5004. [Google Scholar] [CrossRef]
- Magnabosco, C.; Tekere, M.; Lau, M.C.Y.; Linage, B.; Kuloyo, O.; Erasmus, M.; Cason, E.; van Heerden, E.; Borgonie, G.; Kieft, T.L.; et al. Comparisons of the composition and biogeographic distribution of the bacterial communities occupying South African thermal springs with those inhabiting deep subsurface fracture water. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Jonker, C.Z.; van Ginkel, C.; Olivier, J. Association between physical and geochemical characteristics of thermal springs and algal diversity in Limpopo Province, South Africa. Water SA 2013, 39, 95–104. [Google Scholar] [CrossRef]
- Kent, L.E. The thermal waters of the Union of South Africa and South West Africa. S. Afr. J. Geol. 1949, 52, 231–264. [Google Scholar]
- Ackermann, H.-W. Basic phage electron microscopy. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization and Interaction; Humana Press: New York, NY, USA, 2009; pp. 113–126. ISBN 9781603271646. [Google Scholar]
- Hansen, M.C.; Tolker-Nielsen, T.; Givskov, M.; Molin, S. Biased 16S rDNA PCR amplification caused by interference from DNA flanking the template region. FEMS Microbiol. Ecol. 1998, 26, 141–149. [Google Scholar] [CrossRef]
- Reysenbach, A.; Pace, N.; Robb, F.; Place, A. Archaea: A Laboratory Manual—Thermophiles; Cold Spring Harbour Laboratory Press: Cold Spring Harbour, NY, USA, 1995. [Google Scholar]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.E.; Felts, B.; Breitbart, M.; Salamon, P.; Edwards, R.A.; Carlson, C.; Chan, A.M.; Haynes, M.; Kelley, S.; Liu, H.; et al. The marine viromes of four oceanic regions. PLoS Biol. 2006, 4, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.; Rodriguez-Brito, B.; Bangor, D.; McNairnie, P.; Breitbart, M.; Salamon, P.; Felts, B.; Nulton, J.; Mahaffy, J.; Rohwer, F. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinform. 2005, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paez-Espino, D.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Szeto, E.; Pillay, M.; Huang, J.; Markowitz, V.M.; Nielsen, T. IMG/VR: A database of cultured and uncultured DNA Viruses and retroviruses. Nucleic Acids Res. 2017, 45, D457–D465. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 2007, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRcompar: A website to compare clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed]
- Diamond, R.E.; Harris, C. Oxygen and hydrogen isotope geochemistry of thermal springs of the Western Cape, South Africa: Recharge at high altitude? J. Afr. Earth Sci. 2000, 31, 467–481. [Google Scholar] [CrossRef]
- Boekstein, M.S. Revitalising the Healing Tradition-Health Tourism Potential of Thermal Springs in the Western Cape. Thesis (accepted), Cape Peninsula University of Technology, Cape Town, South Africa, 2011. [Google Scholar]
- Mazor, E.; Verhagen, B.T. Dissolved ions, stable and radioactive isotopes and noble gases in thermal waters of South Africa. J. Hydrol. 1983, 63, 315–329. [Google Scholar] [CrossRef]
- Krupovic, M.; Prangishvili, D.; Hendrix, R.W.; Bamford, D.H. Genomics of bacterial and archaeal viruses: Dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 2011, 75, 610–635. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W. Jumbo bacteriophages. Curr. Top. Microbiol. Immunol. 2009, 328, 229–240. [Google Scholar] [PubMed]
- Yuan, Y.; Gao, M. Jumbo bacteriophages: An overview. Front. Microbiol. 2017, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Dyall-Smith, M.; Tang, S.L.; Bath, C. Haloarchaeal viruses: How diverse are they? Res. Microbiol. 2003, 154, 309–313. [Google Scholar] [CrossRef]
- Iverson, E.; Stedman, K. A genetic study of SSV1, the prototypical fusellovirus. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Stedman, K.; Roberto, F.; Willits, D.; Gleske, A.-K.; Zoeller, L.; Snyder, J.; Douglas, T.; Young, M. Comparative genomic analysis of hyperthermophilic archaeal Fuselloviridae viruses. J. Virol. 2004, 78, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Quemin, E.R.J.; Bamford, D.H.; Forterre, P.; Prangishvili, D. Unification of the globally distributed spindle-shaped viruses of the Archaea. J. Virol. 2014, 88, 2354–2358. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lin, J.; Li, N.; Hu, Z.; Deng, F. Characterization and genomic analysis of a plaque purified strain of cyanophage PP. Virol. Sin. 2013, 28, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kong, S.; Shi, M.; Fu, L.; Gao, Y.; An, C. Genomic analysis of freshwater cyanophage Pf-WMP3 infecting cyanobacterium Phormidium foveolarum: The conserved elements for a phage. Microb. Ecol. 2008, 56, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.; Gupta, R.S. Molecular signatures in protein sequences that are characteristics of the phylum Aquificae. Int. J. Syst. Evol. Microbiol. 2006, 56, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Donelli, G.; Dore, E.; Frontali, C.; Grandolfo, M.E. Structure and physico-chemical properties of bacteriophage G. III. A homogeneous DNA of molecular weight 5 × 108. J. Mol. Biol. 1975, 94. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Cowan, D.A. Using signature genes as tools to assess environmental viral ecology and diversity. Appl. Environ. Microbiol. 2014, 80, 4470–4480. [Google Scholar] [CrossRef] [PubMed]
- Prangishvili, D. The wonderful world of archaeal viruses. Annu. Rev. Microbiol. 2013, 67, 565–585. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Pietilä, M.K.; Fu, C.J.; Schmid, M.F.; Bamford, D.H.; Chiu, W. Lemon-shaped halo archaeal virus His1 with uniform tail but variable capsid structure. Proc. Natl. Acad. Sci. USA 2015, 112, 2449–2454. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.K.; Roine, E.; Sencilo, A.; Bamford, D.H.; Oksanen, H.M. Pleolipoviridae, a newly proposed family comprising archaeal pleomorphic viruses with single-stranded or double-stranded DNA genomes. Arch. Virol. 2016, 161, 249–2556. [Google Scholar] [CrossRef]
- Haring, M.; Rachel, R.; Peng, X.; Garrett, R.A.; Prangishvili, D. Viral diversity in hot springs of Pozzuoli, Italy, and characterization of a unique archaeal virus, Acidianus bottle-shaped virus, from a new family, the Ampullaviridae. J. Virol. 2005, 79, 9904–9911. [Google Scholar] [CrossRef] [PubMed]
- Yoosuf, N.; Yutin, N.; Colson, P.; Shabalina, S.A.; Pagnier, I.; Robert, C.; Azza, S.; Klose, T.; Wong, J.; Rossmann, M.G.; et al. Related giant viruses in distant locations and different habitats: Acanthamoeba polyphaga moumouvirus represents a third lineage of the Mimiviridae that is close to the Megavirus lineage. Genome Biol. Evol. 2012, 4, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Bath, C.; Cukalac, T.; Porter, K.; Dyall-Smith, M.L. His1 and His2 are distantly related, spindle-shaped haloviruses belonging to the novel virus group, Salterprovirus. Virology 2006, 350, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Cvirkaite-Krupovic, V.; Iranzo, J.; Prangishvili, D.; Koonin, E.V. Viruses of archaea: Structural, functional, environmental and evolutionary genomics. Virus Res. 2017, 244, 181–193. [Google Scholar] [CrossRef]
- Luk, W.A.; Williams, J.T.; Erdmann, S.; Papke, T.R.; Cavicchioli, R. Viruses of Haloarchaea. Life 2014, 4, 681–715. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wu, G.; Lim, E.S.; Droit, L.; Krishnamurthy, S.; Barouch, D.H.; Virgin, H.W.; Wang, D. VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology 2017, 503, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Kunin, V.; Hugenholtz, P. CRISPR—A widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 2008, 6, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Heidelberg, J.F.; Nelson, W.C.; Schoenfeld, T.; Bhaya, D. Germ warfare in a microbial mat community: CRISPRs provide insights into the co-evolution of host and viral genomes. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Bell, E. Life at Extremes: Environments, Organisms, and Strategies for Survival; CABI: Wallingford, UK, 2012; Volume 1, ISBN 1845938143. [Google Scholar]
- Xia, H.; Li, T.; Deng, F.; Hu, Z. Freshwater cyanophages. Virol. Sin. 2013, 28, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.C.; Goddard, V.J.; Davy, J.; Schroeder, D.C.; Adams, D.G.; Wilson, W.H. Identification of a diagnostic marker to detect freshwater cyanophages of filamentous cyanobacteria. Appl. Environ. Microbiol. 2006, 72, 5713–5719. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.J.; Rusch, D.B.; Yooseph, S.; Halpern, A.L.; Heidelberg, K.B.; Glass, J.I.; Pfannkoch, C.A.; Fadrosh, D.; Miller, C.S.; Sutton, G.; et al. The sorcerer II global ocean sampling expedition: Metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Tekere, M.; Lötter, A.; Olivier, J.; Jonker, N.; Venter, S. Metagenomic analysis of bacterial diversity of Siloam hot water spring, Limpopo, South Africa. Afr. J. Biotechnol. 2011, 10, 18005–18012. [Google Scholar] [CrossRef]
- Aghnatios, R.; Cayrou, C.; Garibal, M.; Robert, C.; Azza, S.; Raoult, D.; Drancourt, M. Draft genome of Gemmata massiliana sp. nov, a water-borne Planctomycetes species exhibiting two variants. Stand. Genom. Sci. 2015, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Senčilo, A.; Jacobs-Sera, D.; Russell, D.A.; Ko, C.-C.; Bowman, C.A.; Atanasova, N.S.; Österlund, E.; Oksanen, H.M.; Bamford, D.H.; Hatfull, G.F.; et al. Snapshot of haloarchaeal tailed virus genomes. RNA Biol. 2013, 10, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, N.S.; Bamford, D.H.; Oksanen, H.M. Haloarchaeal virus morphotypes. Biochimie 2015, 118, 333–343. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Virus Type/Family | No. of Particles | Virion Dimensions (in nm) | |
|---|---|---|---|
| Nucleocapsid Diameter Range | Tail Length | ||
| Myoviridae | 6 | 66–129 | 111–117 |
| Siphoviridae | 51 | 49–111 | 58–318 |
| Podoviridae | 5 | 62–70 | 15–41 |
| Fuselloviridae-like | 8 | 112–160 (40–99) 1 | - |
| Spherical, unknown | 4 | 66–100 | - |
| Virus Family | No. of BLASTp Hits a | Most Represented Isolates |
|---|---|---|
| Myoviridae | 101 | Bacillus virus G |
| Siphoviridae | 23 | Cyanophage PP |
| Podoviridae | 12 | Synechococcus phage S-SKS1 |
| Unclassified | 5 | Prochlorococcus phage Syn1 |
| Domain | Species Name | Sequence Accession Number | Gene Function 1 |
|---|---|---|---|
| Bacteria | Enterobacter sakazakii | NC_009778 | Tail tape measure |
| Luteipulveratus mongoliensis | NZ_CP011112 | Tail tubular protein | |
| Capnocytophaga canimorsus | NC_015846 | Terminase, large | |
| Salmonella enterica | NZ_CP012349 | Unknown | |
| Thermoanaerobacterium thermosaccharolyticum | NC_019970 | Portal protein | |
| Acinetobacter sp. | NC_005966 | Terminase, large | |
| Spirochaeta caldaria | NC_015732 | Phage scaffolding protein | |
| Myxococcus fulvus | NZ_CP006003 | Unknown | |
| Pseudomonas sp. | NZ_CP010892 | Terminase, large | |
| Archaea | Pyrococcus sp. | NC_015474 | Portal protein |
| Thermococcus sp. | NC_016051 | Tail fiber protein | |
| Pyrobaculum sp. | NC_016645 | Terminase, large | |
| Methanothermobacter sp. | NZ_AP011952 | Unknown | |
| Thermococcus piezophilus | NZ_CP015520 | Unknown |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zablocki, O.; Van Zyl, L.J.; Kirby, B.; Trindade, M. Diversity of dsDNA Viruses in a South African Hot Spring Assessed by Metagenomics and Microscopy. Viruses 2017, 9, 348. https://doi.org/10.3390/v9110348
Zablocki O, Van Zyl LJ, Kirby B, Trindade M. Diversity of dsDNA Viruses in a South African Hot Spring Assessed by Metagenomics and Microscopy. Viruses. 2017; 9(11):348. https://doi.org/10.3390/v9110348
Chicago/Turabian StyleZablocki, Olivier, Leonardo Joaquim Van Zyl, Bronwyn Kirby, and Marla Trindade. 2017. "Diversity of dsDNA Viruses in a South African Hot Spring Assessed by Metagenomics and Microscopy" Viruses 9, no. 11: 348. https://doi.org/10.3390/v9110348
APA StyleZablocki, O., Van Zyl, L. J., Kirby, B., & Trindade, M. (2017). Diversity of dsDNA Viruses in a South African Hot Spring Assessed by Metagenomics and Microscopy. Viruses, 9(11), 348. https://doi.org/10.3390/v9110348