Implication of Different HIV-1 Genes in the Modulation of Autophagy


Abstract
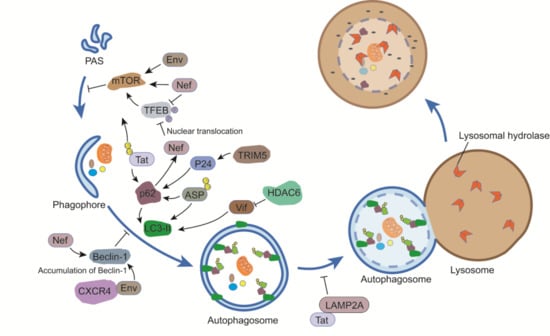
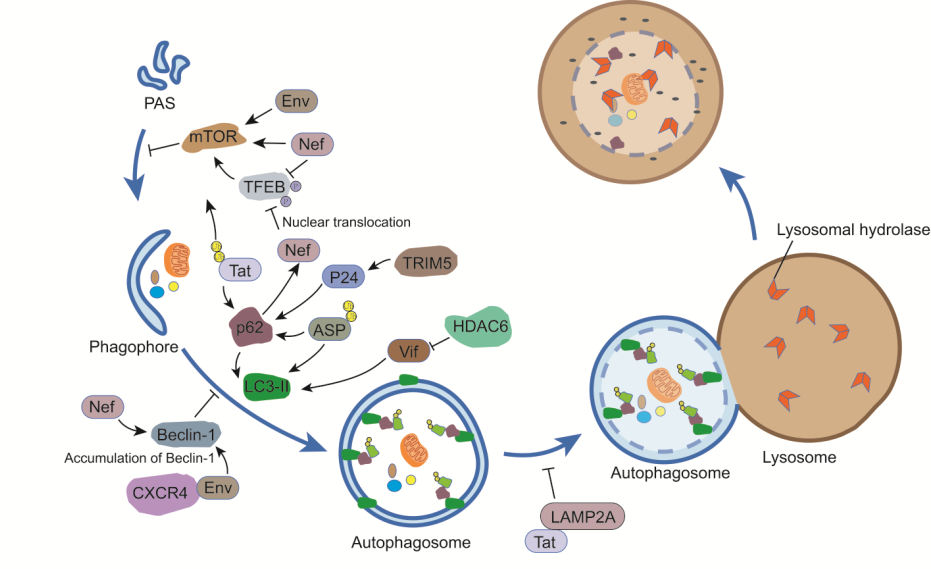
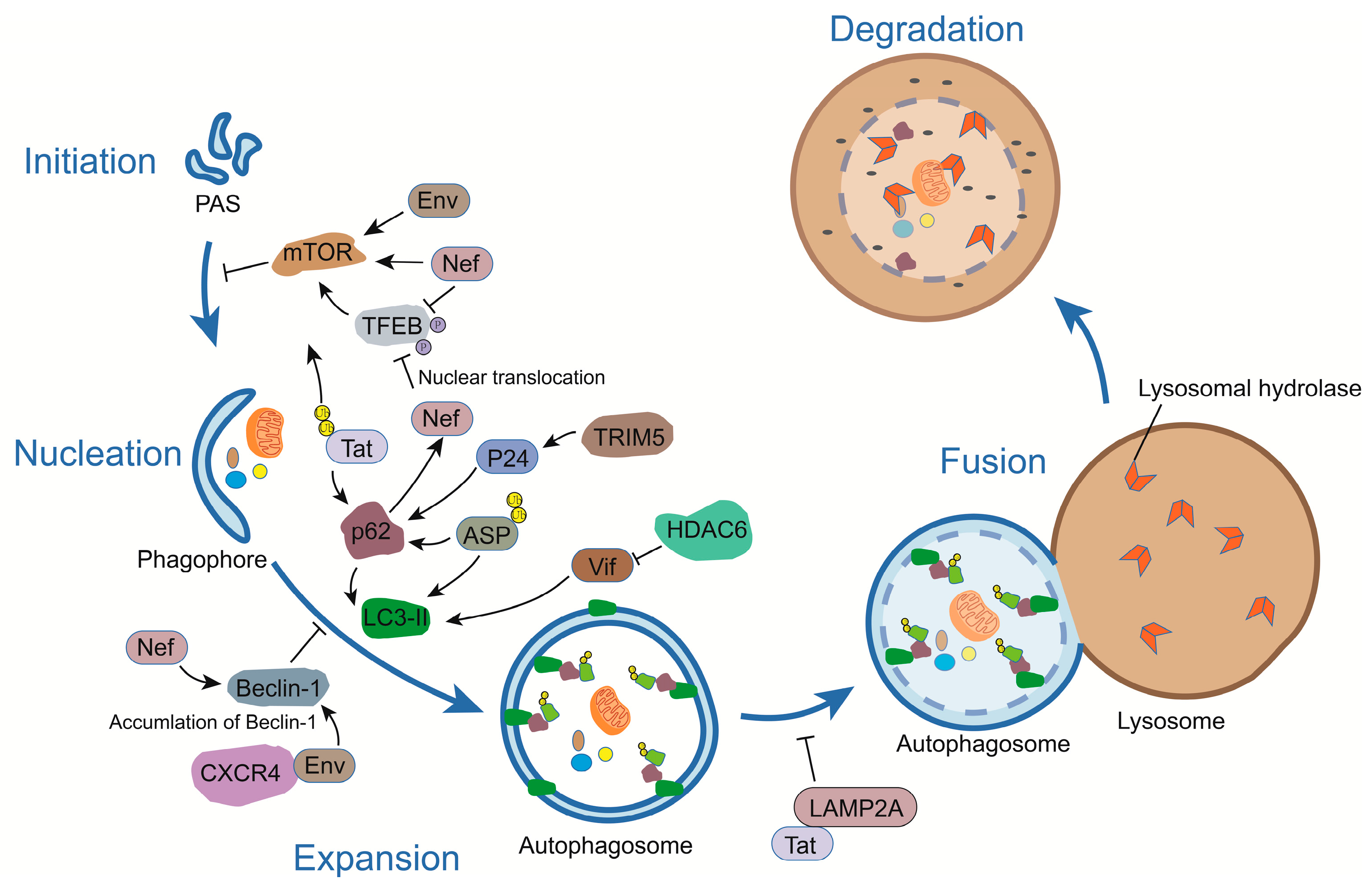
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
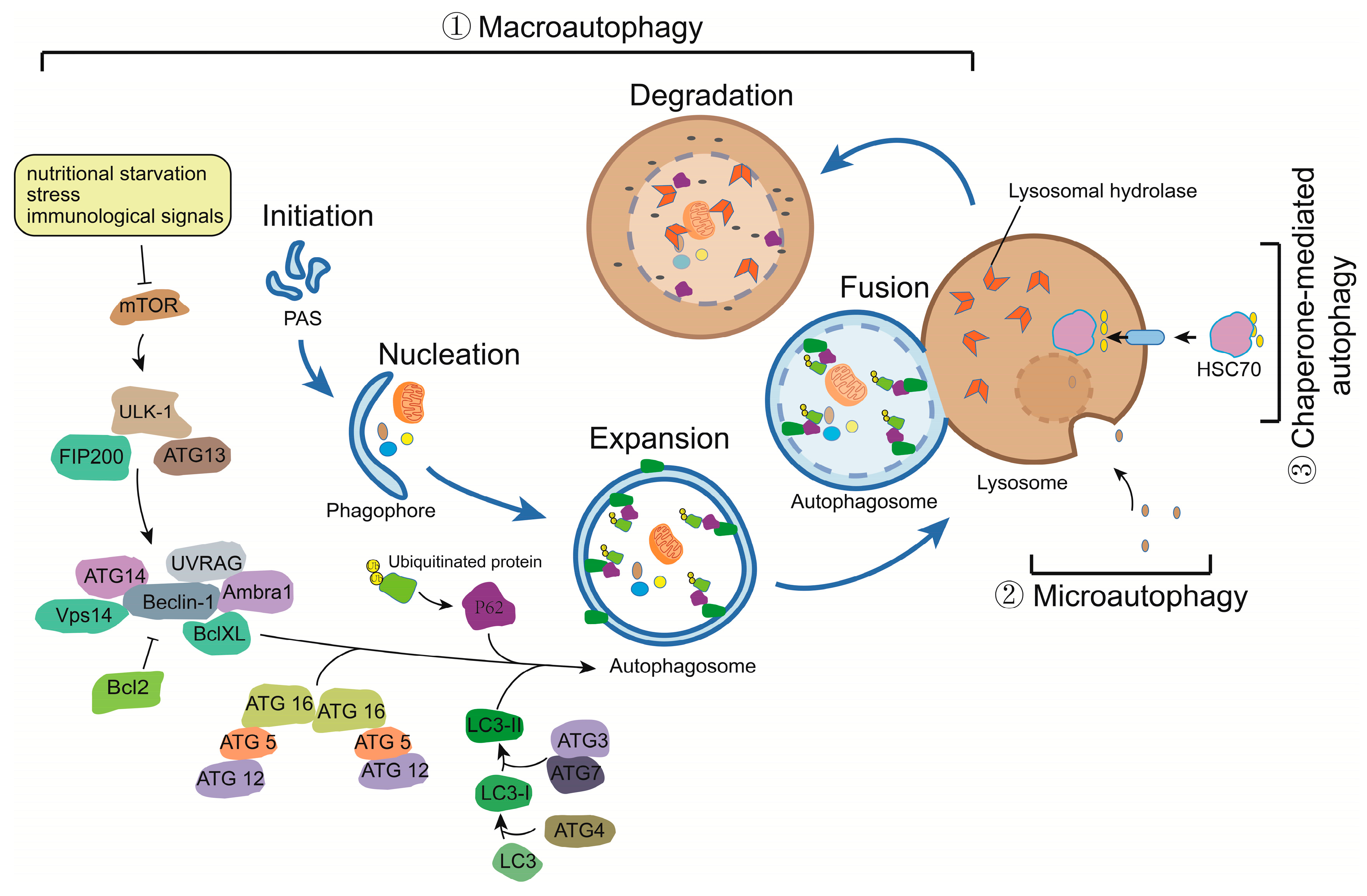
2. The Molecular Machinery of Autophagy
3. A Crosstalk between Autophagy and HIV-1
3.1. Env
3.2. Gag
3.3. Tat
3.4. Nef
3.5. Vif
3.6. Vpu
3.7. ASP
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jia, J.; Rodrigues, B. Autophagy, metabolic disease, and pathogenesis of heart dysfunction. Can. J. Cardiol. 2017, 33, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Vural, A.; Kehrl, J.H. Autophagy in macrophages: Impacting inflammation and bacterial infection. Scientifica 2014, 2014, 825463. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [PubMed]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Ciccosanti, F.; Marsella, C.; Ippolito, G.; Piacentini, M.; Fimia, G.M. Role of autophagy in HIV infection and pathogenesis. J. Intern. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Patgaonkar, M.; Shroff, A.; Ayyar, K.; Bashir, T.; Reddy, K.V. Hemoglobin expression in nonerythroid cells: Novel or ubiquitous? Int. J. Inflamm. 2014, 2014, 803237. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Findlay, G.M.; Yan, L.; Procter, J.; Mieulet, V.; Lamb, R.F. A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem. J. 2007, 403, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Nazarko, V.Y.; Zhong, Q. ULK1 targets Beclin-1 in autophagy. Nat. Cell Biol. 2013, 15, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, K.H. Autophagy: Kinase crosstalk through Beclin 1. Nat. Rev. Mol. Cell Biol. 2013, 14, 402–403. [Google Scholar] [CrossRef] [PubMed]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Codogno, P.; Morel, E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017, 284, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim. Biophys. Acta 2013, 1833, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, G.; Martens, S. Mechanisms of selective autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed. Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell 2013, 51, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Glazyrin, A.; Kumar, S.; Kumar, A. Role of autophagy in HIV pathogenesis and drug abuse. Mol. Neurobiol. 2016, 54, 5855–5867. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Beaumelle, B.; Vergne, I. Autophagy in Mycobacterium tuberculosis and HIV infections. Front. Cell. Infect. Microbiol. 2015, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef] [PubMed]
- Perfettini, J.L.; Castedo, M.; Roumier, T.; Andreau, K.; Nardacci, R.; Piacentini, M.; Kroemer, G. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005, 12 (Suppl. 1), 916–923. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Denizot, M.; Varbanov, M.; Espert, L.; Robert-Hebmann, V.; Sagnier, S.; Garcia, E.; Curriu, M.; Mamoun, R.; Blanco, J.; Biard-Piechaczyk, M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 2008, 4, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 2009, 4, e5787. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Masliah, E.; Spector, S.A. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J. Infect. Dis. 2011, 203, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM family proteins: Roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Mandell, M.A.; Kimura, T.; Jain, A.; Johansen, T.; Deretic, V. TRIM proteins regulate autophagy: TRIM5 is a selective autophagy receptor mediating HIV-1 restriction. Autophagy 2014, 10, 2387–2388. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Towers, G.J.; Qasim, W. Gene therapy strategies to exploit TRIM derived restriction factors against HIV-1. Viruses 2014, 6, 243–263. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.; Pertel, T.; Gray, S.; Robia, S.L.; Bakowska, J.C.; Luban, J.; Campbell, E.M. p62/sequestosome-1 associates with and sustains the expression of retroviral restriction factor TRIM5alpha. J. Virol. 2010, 84, 5997–6006. [Google Scholar] [CrossRef] [PubMed]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Munch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Shi, H.X.; Liu, X.Y.; Shan, Y.F.; Wei, B.; Chen, S.; Wang, C. TRIM21 is essential to sustain IFN regulatory factor 3 activation during antiviral response. J. Immunol. 2009, 182, 3782–3792. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Harwig, A.; Berkhout, B. The HIV-1 Tat protein has a versatile role in activating viral transcription. J. Virol. 2011, 85, 9506–9516. [Google Scholar] [CrossRef] [PubMed]
- Kamori, D.; Ueno, T. HIV-1 Tat and viral latency: What we can learn from naturally occurring sequence variations. Front. Microbiol. 2017, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Bres, V.; Kiernan, R.E.; Linares, L.K.; Chable-Bessia, C.; Plechakova, O.; Treand, C.; Emiliani, S.; Peloponese, J.M.; Jeang, K.T.; Coux, O.; et al. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 2003, 5, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qin, J.; Li, Y.; Wang, J.; He, Q.; Zhou, J.; Liu, M.; Li, D. Modulation of the stability and activities of HIV-1 Tat by its ubiquitination and carboxyl-terminal region. Cell Biosci. 2014, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.; Dumaop, W.; Eleuteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: Implications for HIV-associated neurocognitive disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, E.N.; Dikeakos, J.D. HIV-1 Nef: A master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim. Biophys. Acta 2015, 1850, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human immunodeficiency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [PubMed]
- Sardo, L.; Iordanskiy, S.; Klase, Z.; Kashanchi, F. HIV-1 Nef blocks autophagy in human astrocytes. Cell Cycle 2015, 14, 3781–3782. [Google Scholar] [CrossRef] [PubMed]
- Saribas, A.S.; Khalili, K.; Sariyer, I.K. Dysregulation of autophagy by HIV-1 Nef in human astrocytes. Cell Cycle 2015, 14, 2899–2904. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.M.; Marin, M.; Kozak, S. L.; Kabat, D. The viral infectivity factor (Vif) of HIV-1 unveiled. Trends Mol. Med. 2004, 10, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Iwatani, Y. APOBEC3G-mediated G-to-A hypermutation of the HIV-1 genome: The missing link in antiviral molecular mechanisms. Front. Microbiol. 2016, 7, 2027. [Google Scholar] [CrossRef] [PubMed]
- Borel, S.; Robert-Hebmann, V.; Alfaisal, J.; Jain, A.; Faure, M.; Espert, L.; Chaloin, L.; Paillart, J.C.; Johansen, T.; Biard-Piechaczyk, M. HIV-1 viral infectivity factor interacts with microtubule-associated protein light chain 3 and inhibits autophagy. AIDS 2015, 29, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Valera, M.S.; de Armas-Rillo, L.; Barroso-Gonzalez, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernandez, S.; Borel, S.; Garcia-Exposito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Madjo, U.; Leymarie, O.; Fremont, S.; Kuster, A.; Nehlich, M.; Gallois-Montbrun, S.; Janvier, K.; Berlioz-Torrent, C. LC3C contributes to Vpu-mediated antagonism of BST2/Tetherin restriction on HIV-1 release through a non-canonical autophagy pathway. Cell Rep. 2016, 17, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Cassan, E.; Arigon-Chifolleau, A.M.; Mesnard, J.M.; Gross, A.; Gascuel, O. Concomitant emergence of the Antisense Protein gene of HIV-1 and of the pandemic. Proc. Natl. Acad. Sci. USA 2016, 113, 11537–11542. [Google Scholar] [CrossRef] [PubMed]
- Landry, S.; Halin, M.; Lefort, S.; Audet, B.; Vaquero, C.; Mesnard, J.M.; Barbeau, B. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology 2007, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Laverdure, S.; Gross, A.; Arpin-Andre, C.; Clerc, I.; Beaumelle, B.; Barbeau, B.; Mesnard, J.M. HIV-1 antisense transcription is preferentially activated in primary monocyte-derived cells. J. Virol. 2012, 86, 13785–13789. [Google Scholar] [CrossRef] [PubMed]
- Torresilla, C.; Larocque, E.; Landry, S.; Halin, M.; Coulombe, Y.; Masson, J.Y.; Mesnard, J.M.; Barbeau, B. Detection of the HIV-1 minus-strand-encoded Antisense Protein and its association with autophagy. J. Virol. 2013, 87, 5089–5105. [Google Scholar] [CrossRef] [PubMed]
- Bet, A.; Maze, E.A.; Bansal, A.; Sterrett, S.; Gross, A.; Graff-Dubois, S.; Samri, A.; Guihot, A.; Katlama, C.; Theodorou, I.; et al. The HIV-1 Antisense Protein (ASP) induces CD8 T cell responses during chronic infection. Retrovirology 2015, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Vanhee-Brossollet, C.; Thoreau, H.; Serpente, N.; D’Auriol, L.; Levy, J.P.; Vaquero, C. A natural antisense RNA derived from the HIV-1 Env gene encodes a protein which is recognized by circulating antibodies of HIV+ individuals. Virology 1995, 206, 196–202. [Google Scholar] [CrossRef]
- Bansal, A.; Carlson, J.; Yan, J.; Akinsiku, O.T.; Schaefer, M.; Sabbaj, S.; Bet, A.; Levy, D.N.; Heath, S.; Tang, J. CD8 T cell response and evolutionary pressure to HIV-1 cryptic epitopes derived from antisense transcription. J. Exp. Med. 2010, 207, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, C.T.; Llano, A.; Carlson, J.M.; Brumme, Z.L.; Brockman, M.A.; Cedeno, S.; Harrigan, P.R.; Kaufmann, D.E.; Heckerman, D.; Meyerhans, A.; et al. Immune screening identifies novel T cell targets encoded by anti-sense reading frames of HIV-1. J. Virol. 2015, 89, 4015–4019. [Google Scholar] [CrossRef] [PubMed]
- Briquet, S.; Vaquero, C. Immunolocalization studies of an Antisense Protein in HIV-1-infected cells and viral particles. Virology 2002, 292, 177–184. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Xiao, Y.; Torresilla, C.; Rassart, É.; Barbeau, B. Implication of Different HIV-1 Genes in the Modulation of Autophagy. Viruses 2017, 9, 389. https://doi.org/10.3390/v9120389
Liu Z, Xiao Y, Torresilla C, Rassart É, Barbeau B. Implication of Different HIV-1 Genes in the Modulation of Autophagy. Viruses. 2017; 9(12):389. https://doi.org/10.3390/v9120389
Chicago/Turabian StyleLiu, Zhenlong, Yong Xiao, Cynthia Torresilla, Éric Rassart, and Benoit Barbeau. 2017. "Implication of Different HIV-1 Genes in the Modulation of Autophagy" Viruses 9, no. 12: 389. https://doi.org/10.3390/v9120389
APA StyleLiu, Z., Xiao, Y., Torresilla, C., Rassart, É., & Barbeau, B. (2017). Implication of Different HIV-1 Genes in the Modulation of Autophagy. Viruses, 9(12), 389. https://doi.org/10.3390/v9120389