Myxoma Virus dsRNA Binding Protein M029 Inhibits the Type I IFN‐Induced Antiviral State in a Highly Species‐Specific Fashion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Construction of Recombinant Viruses and Viral Preparation
2.3. Interferon (IFN) Sensitivity and Virus Replication Assays
2.4. Luciferase Assay
2.5. RNA Purification and Real-Time Polymerase Chain Reaction (qPCR)
2.6. Western Blot Analysis
2.7. Generation of Knockout Cells Using CRISPR/Cas9
2.8. Statistical Analysis
3. Results
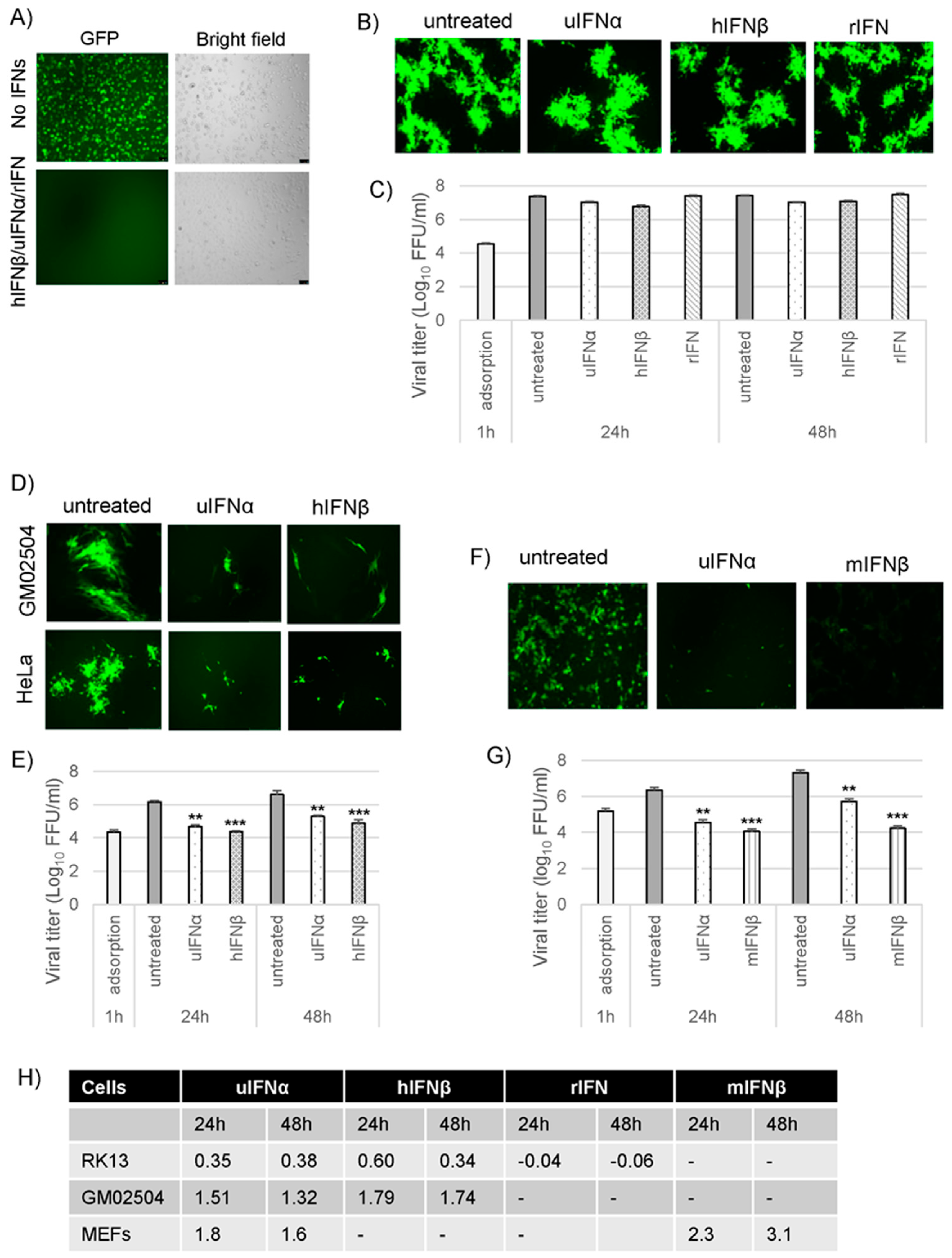
3.1. Species-Specific Inhibition of Type I IFN-Induced Antiviral States by Myxoma Virus (MYXV)
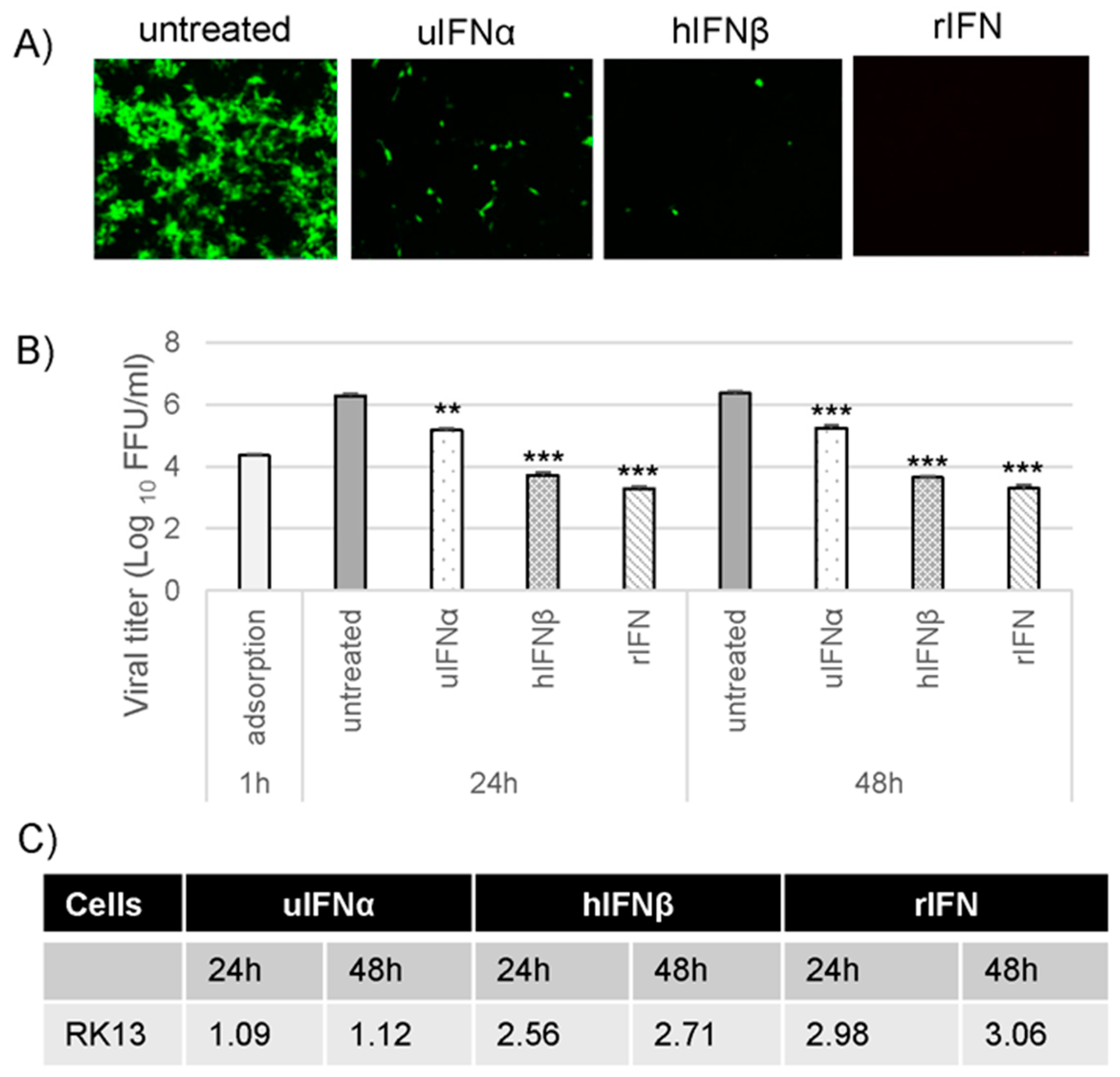
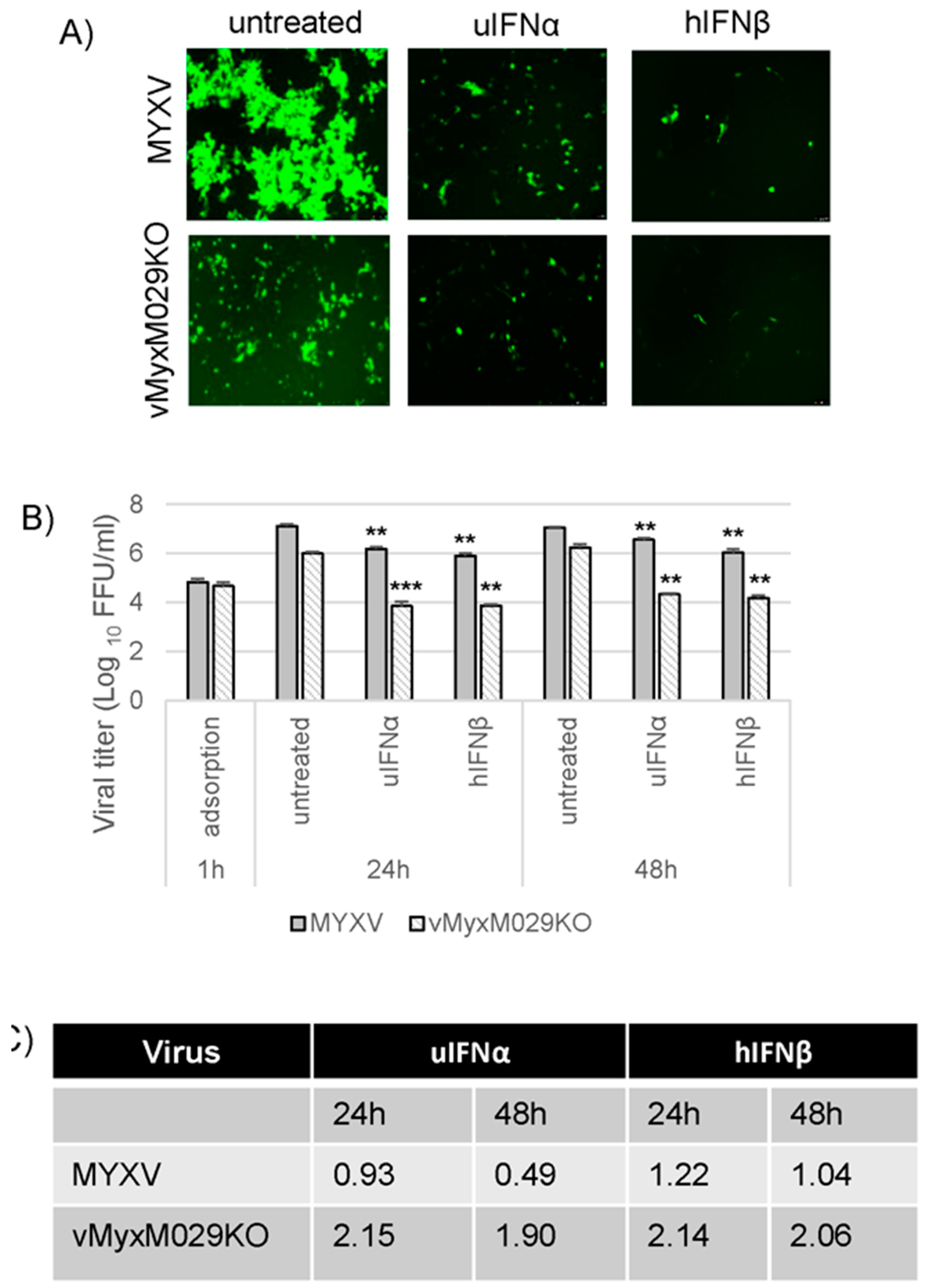
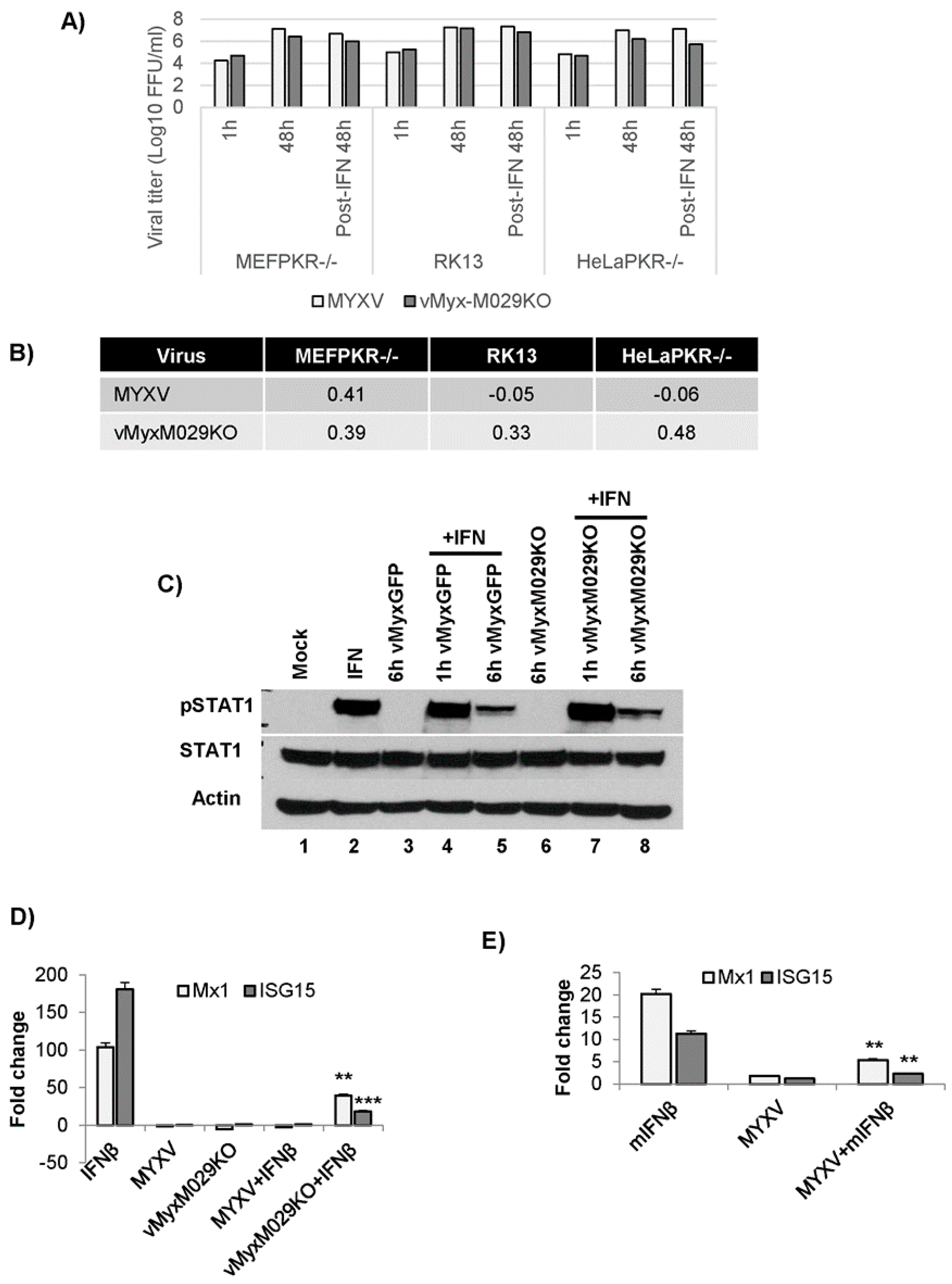
3.2. M029 is Required for the Global Inhibition of Type I IFN-Induced Antiviral States in Rabbit Cells
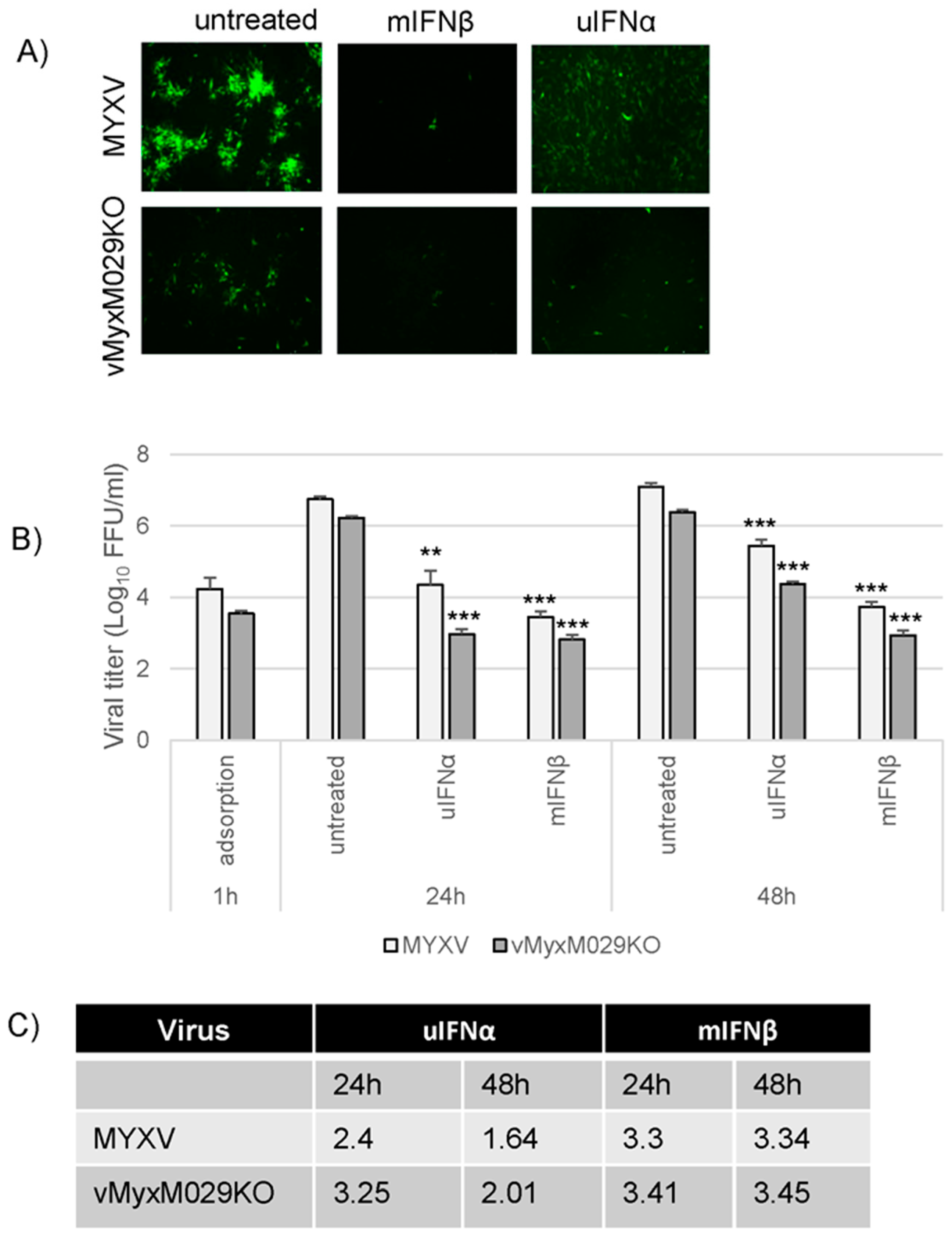
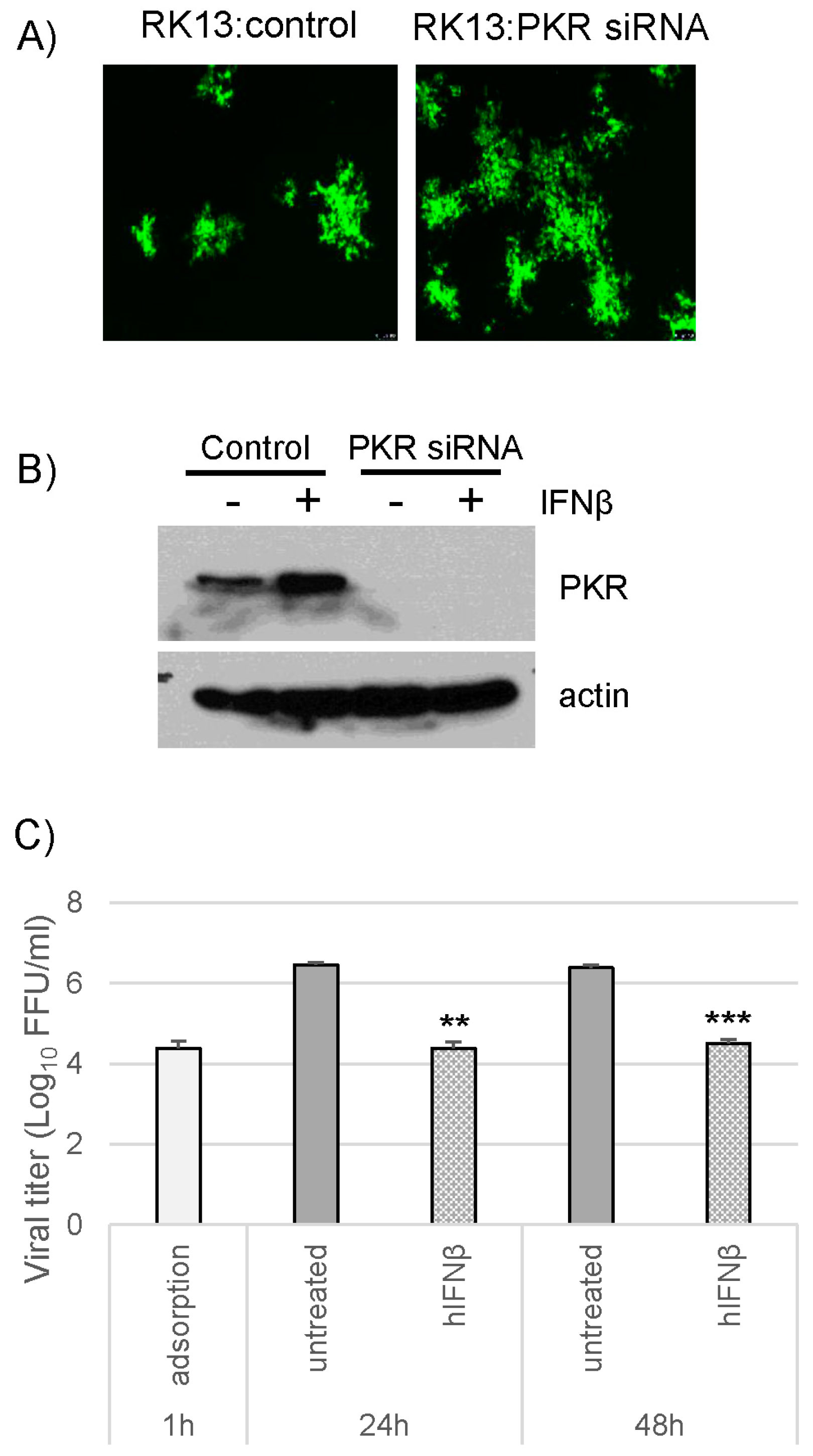
3.3. Loss of Protein Kinase R (PKR) Can Rescue M029-Minus MYXV Replication in Mouse Embryo Fibroblasts (MEFs) but Is Unable to Rescue Virus Resistance to the Mouse Type I IFN-Induced Antiviral State
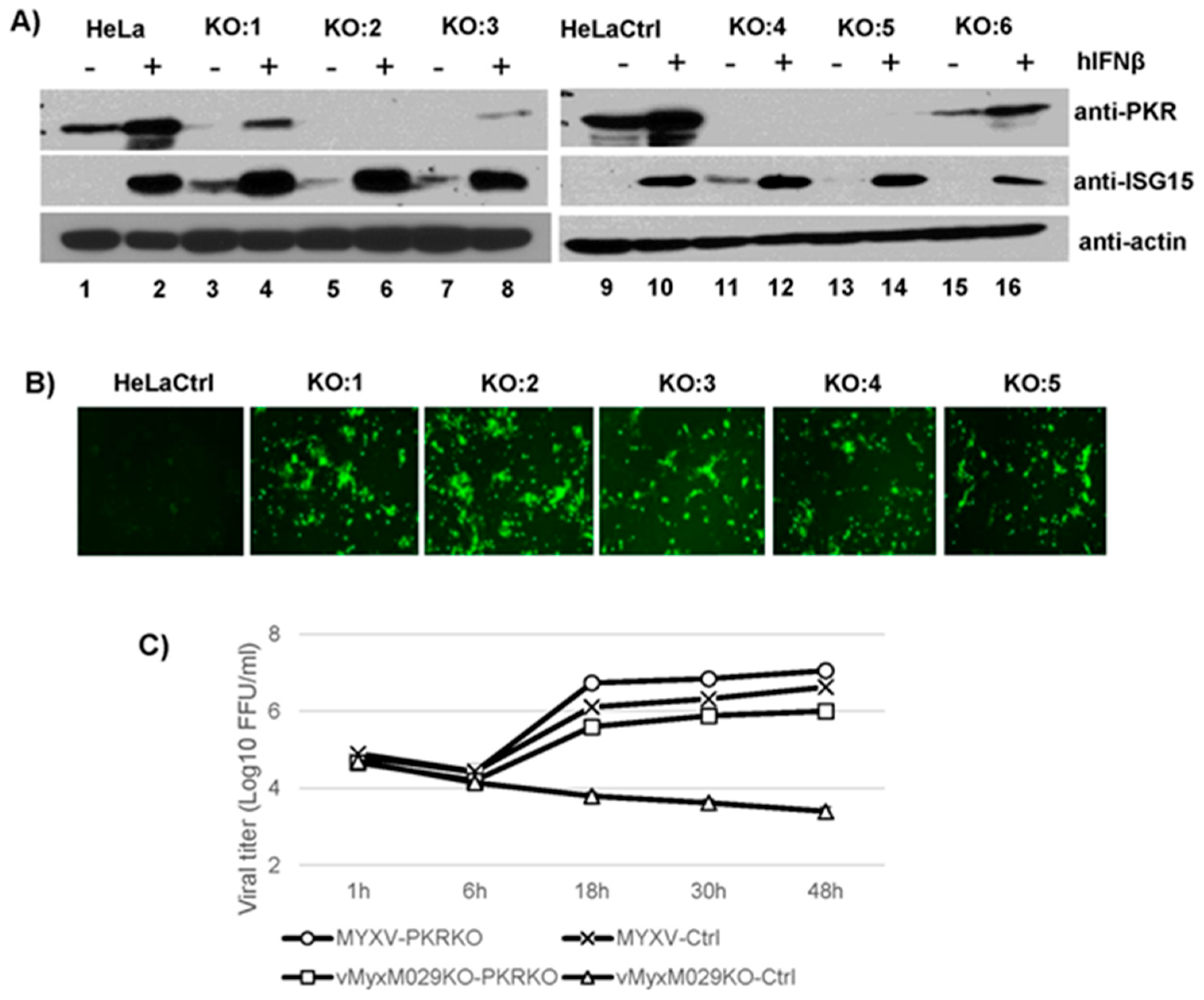
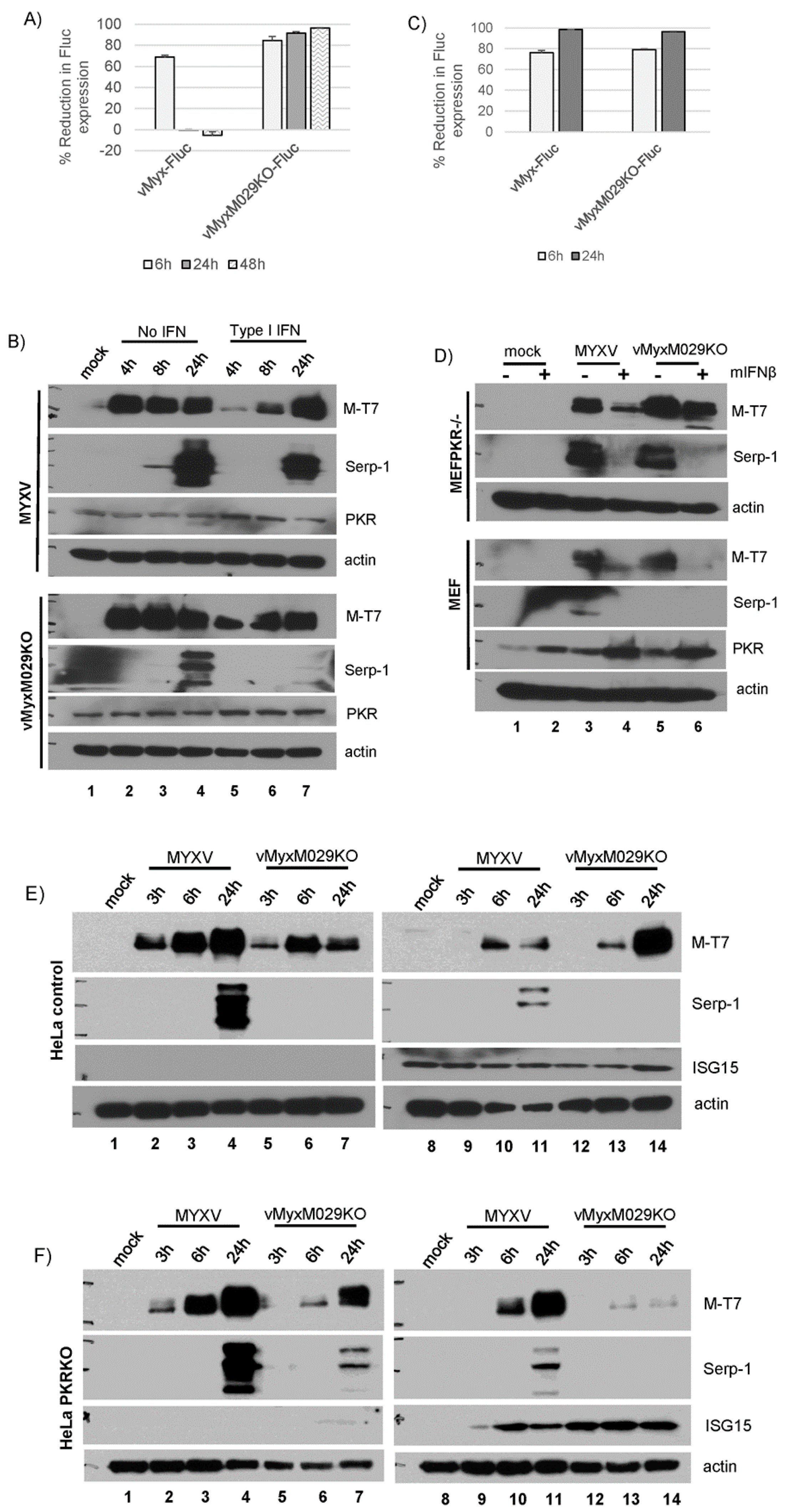
3.4. M029 is Required for the Partial Inhibition of Type I IFN-Induced Antiviral States in Human Cells in Either the Presence or Absence of PKR
3.5. PKR Knockdown in Rabbit Cells Cannot Rescue vMyxM029KO Virus Replication after Establishment of the Type I IFN-Induced Antiviral State
3.6. Type I IFN-Induced Antiviral States Inhibit Viral Protein Synthesis in a Highly Species-Specific Manner
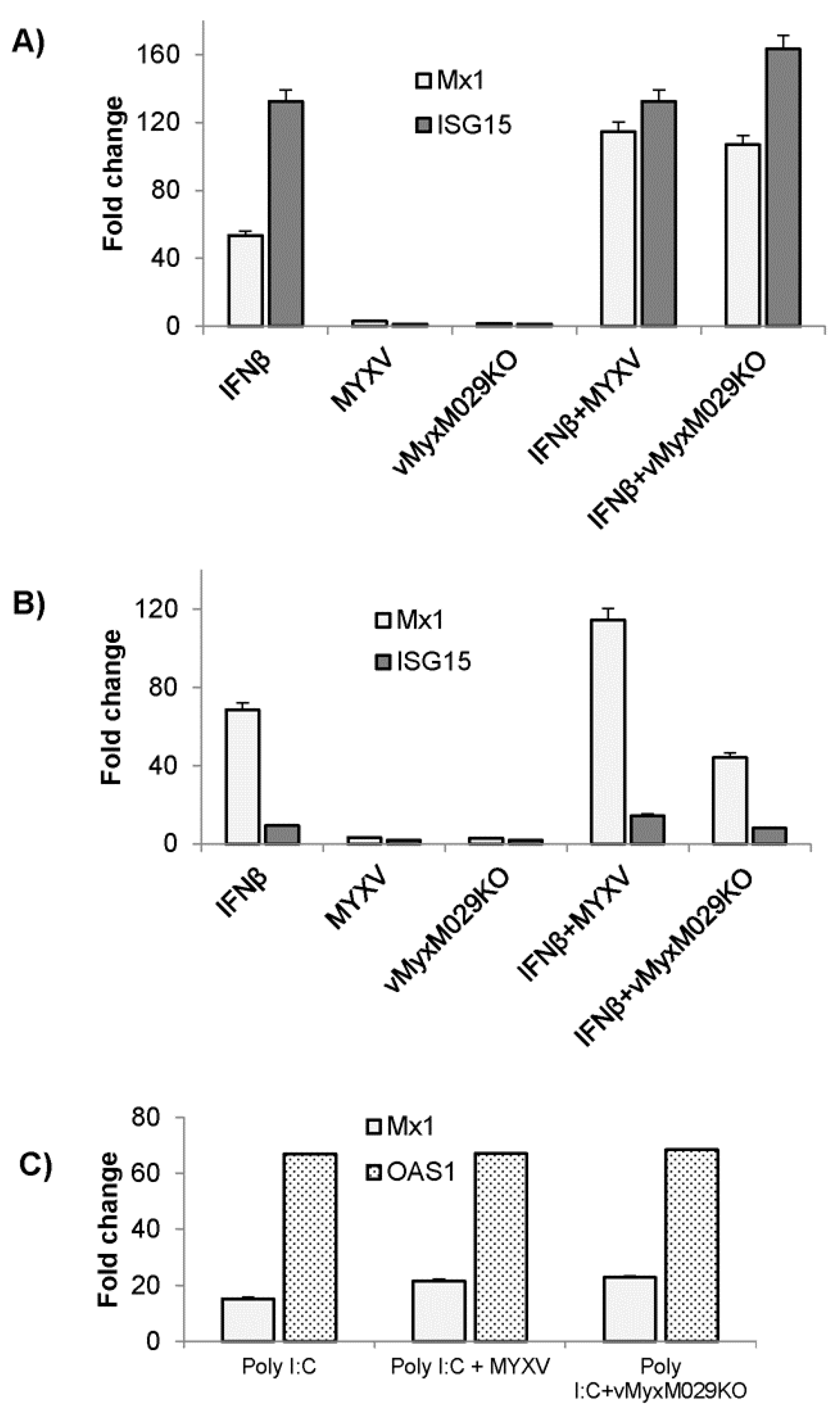
3.7. MYXV Does Not Alter the Levels of Pre-Existing Type I IFN-Induced IFN-Stimulated Genes (ISGs) in Rabbit, Human or Mouse Cells
3.8. MYXV Can Inhibit Type I IFN Signaling Even in the Absence of M029 in Multiple Cell Species
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoffmann, H.H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Stifter, S.A.; Feng, C.G. Interfering with immunity: Detrimental role of type I IFNs during infection. J. Immunol. 2015, 194, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Kerr, P.J.; Ghedin, E.; DePasse, J.V.; Fitch, A.; Cattadori, I.M.; Hudson, P.J.; Tscharke, D.C.; Read, A.F.; Holmes, E.C. Evolutionary history and attenuation of myxoma virus on two continents. PLoS Pathog. 2013, 8, e1002950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, P.J.; Liu, J.; Cattadori, I.; Ghedin, E.; Read, A.F.; Holmes, E.C. Myxoma virus and the Leporipoxviruses: An evolutionary paradigm. Viruses 2015, 7, 1020–1061. [Google Scholar] [CrossRef] [PubMed]
- Werden, S.J.; Rahman, M.M.; McFadden, G. Poxvirus host range genes. Adv. Virus Res. 2008, 71, 135–171. [Google Scholar] [PubMed]
- Bartee, E.; Mohamed, M.R.; Lopez, M.C.; Baker, H.V.; McFadden, G. The addition of tumor necrosis factor plus beta interferon induces a novel synergistic antiviral state against poxviruses in primary human fibroblasts. J. Virol. 2009, 83, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, Y.; Barrett, J.W.; Gao, X.; Loh, J.; Barton, E.; Virgin, H.W.; McFadden, G. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat. Immunol. 2004, 5, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Barrett, J.W.; Ma, Y.; Dekaban, G.A.; McFadden, G. Induction of alpha/beta interferon by myxoma virus is selectively abrogated when primary mouse embryo fibroblasts become immortalized. J. Virol. 2009, 83, 5928–5932. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Gao, X.; Barrett, J.W.; Shao, Q.; Bartee, E.; Mohamed, M.R.; Rahman, M.; Werden, S.; Irvine, T.; Cao, J.; et al. RIG-I mediates the co-induction of tumor necrosis factor and type I interferon elicited by myxoma virus in primary human macrophages. PLoS Pathog. 2008, 4, e1000099. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.M.; McFadden, G. Oncolytic Poxviruses. Annu. Rev. Virol. 2014, 1, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wennier, S.; McFadden, G. The immunoregulatory properties of oncolytic myxoma virus and their implications in therapeutics. Microbes Infect. 2010, 12, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Chan, W. M.; Rahman, M. M.; McFadden, G. Oncolytic myxoma virus: The path to clinic. Vaccine 2013, 31, 4252–4258. [Google Scholar] [CrossRef] [PubMed]
- Seet, B.T.; Johnston, J.B.; Brunetti, C.R.; Barrett, J.W.; Everett, H.; Cameron, C.; Sypula, J.; Nazarian, S.H.; Lucas, A.; McFadden, G. Poxviruses and immune evasion. Annu. Rev. Immunol. 2003, 21, 377–423. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.; Bowie, A.G. Innate immune activation of NFκB and its antagonism by poxviruses. Cytokine Growth Factor Rev. 2014, 25, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, B.; Esteban, M. The interferon system and vaccinia virus evasion mechanisms. J. Interferon Cytokine Res. 2009, 29, 581–598. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Benfield, C.T.; Maluquer de Motes, C.; Mazzon, M.; Ember, S.W.; Ferguson, B.J.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.; Kauffman, E.B.; Martinez, H.; Perkus, M.E.; Jacobs, B.L.; Paoletti, E.; Tartaglia, J. Host-range restriction of vaccinia virus E3L-specific deletion mutants. Virus Genes 1996, 12, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Perkus, M.E.; Goebel, S.J.; Davis, S.W.; Johnson, G.P.; Limbach, K.; Norton, E.K.; Paoletti, E. Vaccinia virus host range genes. Virology 1990, 179, 276–286. [Google Scholar] [CrossRef]
- Meng, X.; Schoggins, J.; Rose, L.; Cao, J.; Ploss, A.; Rice, C. M.; Xiang, Y. C7L family of poxvirus host range genes inhibits antiviral activities induced by type I interferons and interferon regulatory factor 1. J. Virol. 2012, 86, 4538–4547. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Marie, I.; Prakash, A.; Garcia-Sastre, A.; Levy, D.E. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or IκB kinase but is blocked by Vaccinia virus E3L protein. J. Biol. Chem. 2001, 276, 8951–8957. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Condit, R. C.; Vijaysri, S.; Jacobs, B.; Williams, B.R.; Silverman, R.H. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 2002, 76, 5251–5259. [Google Scholar] [CrossRef] [PubMed]
- Myskiw, C.; Arsenio, J.; van Bruggen, R.; Deschambault, Y.; Cao, J. Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-κB, and IRF3 pathways. J. Virol. 2009, 83, 6757–6768. [Google Scholar] [CrossRef] [PubMed]
- Guerra, S.; Caceres, A.; Knobeloch, K. P.; Horak, I.; Esteban, M. Vaccinia virus E3 protein prevents the antiviral action of ISG15. PLoS Pathog. 2008, 4, e1000096. [Google Scholar] [CrossRef] [PubMed]
- Langland, J.O.; Jacobs, B.L. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology 2002, 299, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Myskiw, C.; Arsenio, J.; Hammett, C.; van Bruggen, R.; Deschambault, Y.; Beausoleil, N.; Babiuk, S.; Cao, J. Comparative analysis of poxvirus orthologues of the vaccinia virus E3 protein: Modulation of protein kinase R activity, cytokine responses, and virus pathogenicity. J. Virol. 2011, 85, 12280–12291. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Liu, J.; Chan, W.M.; Rothenburg, S.; McFadden, G. Myxoma virus protein M029 is a dual function immunomodulator that inhibits PKR and also conscripts RHA/DHX9 to promote expanded host tropism and viral replication. PLoS Pathog. 2013, 9, e1003465. [Google Scholar] [CrossRef] [PubMed]
- White, S.D.; Jacobs, B.L. The amino terminus of the vaccinia virus E3 protein is necessary to inhibit the interferon response. J. Virol. 2012, 86, 5895–5904. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; Barrett, J.W.; Chang, W.; Chung, C.S.; Zeng, W.; Masters, J.; Mann, M.; Wang, F.; Cao, J.; McFadden, G. Role of the serine-threonine kinase PAK-1 in myxoma virus replication. J. Virol. 2003, 77, 5877–5888. [Google Scholar] [CrossRef] [PubMed]
- Zemp, F.J.; Lun, X.; McKenzie, B.A.; Zhou, H.; Maxwell, L.; Sun, B.; Kelly, J.J.; Stechishin, O.; Luchman, A.; Weiss, S.; et al. Treating brain tumor-initiating cells using a combination of myxoma virus and rapamycin. Neuro Oncol. 2013, 15, 904–920. [Google Scholar] [CrossRef] [PubMed]
- Everett, H.; Barry, M.; Sun, X.; Lee, S.F.; Frantz, C.; Berthiaume, L.G.; McFadden, G.; Bleackley, R.C. The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore. J. Exp. Med. 2002, 196, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.W.; Sypula, J.; Wang, F.; Alston, L.R.; Shao, Z.; Gao, X.; Irvine, T.S.; McFadden, G. M135R is a novel cell surface virulence factor of myxoma virus. J. Virol. 2007, 81, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Mossman, K.; Nation, P.; Macen, J.; Garbutt, M.; Lucas, A.; McFadden, G. Myxoma virus M-T7, a secreted homolog of the interferon-gamma receptor, is a critical virulence factor for the development of myxomatosis in European rabbits. Virology 1996, 215, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Nash, P.; Barry, M.; Seet, B.T.; Veugelers, K.; Hota, S.; Heger, J.; Hodgkinson, C.; Graham, K.; Jackson, R.J.; McFadden, G. Post-translational modification of the myxoma-virus anti-inflammatory serpin SERP-1 by a virally encoded sialyltransferase. Biochem. J. 2000, 347, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Waibler, Z.; Anzaghe, M.; Frenz, T.; Schwantes, A.; Pohlmann, C.; Ludwig, H.; Palomo-Otero, M.; Alcami, A.; Sutter, G.; Kalinke, U. Vaccinia virus-mediated inhibition of type I interferon responses is a multifactorial process involving the soluble type I interferon receptor B18 and intracellular components. J. Virol. 2009, 83, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Haller, S.L.; Rahman, M.M.; McFadden, G.; Rothenburg, S. Myxoma virus M156 is a specific inhibitor of rabbit PKR but contains a loss-of-function mutation in Australian virus isolates. Proc. Natl. Acad. Sci. USA 2016, 113, 3855–3860. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Barrett, J.W.; Shao, Q.; Gao, X.; Dekaban, G.A.; McFadden, G. Myxoma virus selectively disrupts type I interferon signaling in primary human fibroblasts by blocking the activation of the Janus kinase Tyk2. Virology 2009, 387, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.A.; Huang, J.H.; Li, P.; Chang, H.C.; Slee, R.B.; O’Sullivan, A.; Anita, M.; Yeh, N.; Klemsz, M.J.; Brutkiewicz, R.R.; et al. Vaccinia virus blocks Stat1-dependent and Stat1-independent gene expression induced by type I and type II interferons. J. Interferon Cytokine Res. 2008, 28, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Najarro, P.; Traktman, P.; Lewis, J.A. Vaccinia virus blocks gamma interferon signal transduction: Viral VH1 phosphatase reverses Stat1 activation. J. Virol. 2001, 75, 3185–3196. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.; Tartaglia, J.; Paoletti, E. Vaccinia virus-encoded eIF-2α homolog abrogates the antiviral effect of interferon. Virology 1991, 183, 419–422. [Google Scholar] [CrossRef]
- Unterholzner, L.; Sumner, R.P.; Baran, M.; Ren, H.; Mansur, D.S.; Bourke, N.M.; Randow, F.; Smith, G.L.; Bowie, A.G. Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog. 2011, 7, e1002247. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.M.; McFadden, G. Myxoma Virus dsRNA Binding Protein M029 Inhibits the Type I IFN‐Induced Antiviral State in a Highly Species‐Specific Fashion. Viruses 2017, 9, 27. https://doi.org/10.3390/v9020027
Rahman MM, McFadden G. Myxoma Virus dsRNA Binding Protein M029 Inhibits the Type I IFN‐Induced Antiviral State in a Highly Species‐Specific Fashion. Viruses. 2017; 9(2):27. https://doi.org/10.3390/v9020027
Chicago/Turabian StyleRahman, Masmudur M., and Grant McFadden. 2017. "Myxoma Virus dsRNA Binding Protein M029 Inhibits the Type I IFN‐Induced Antiviral State in a Highly Species‐Specific Fashion" Viruses 9, no. 2: 27. https://doi.org/10.3390/v9020027
APA StyleRahman, M. M., & McFadden, G. (2017). Myxoma Virus dsRNA Binding Protein M029 Inhibits the Type I IFN‐Induced Antiviral State in a Highly Species‐Specific Fashion. Viruses, 9(2), 27. https://doi.org/10.3390/v9020027