
Porcine Epidemic Diarrhea Virus Induces Autophagy to Benefit Its Replication

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Virus Infection
2.3. Quantitative Real-Time PCR
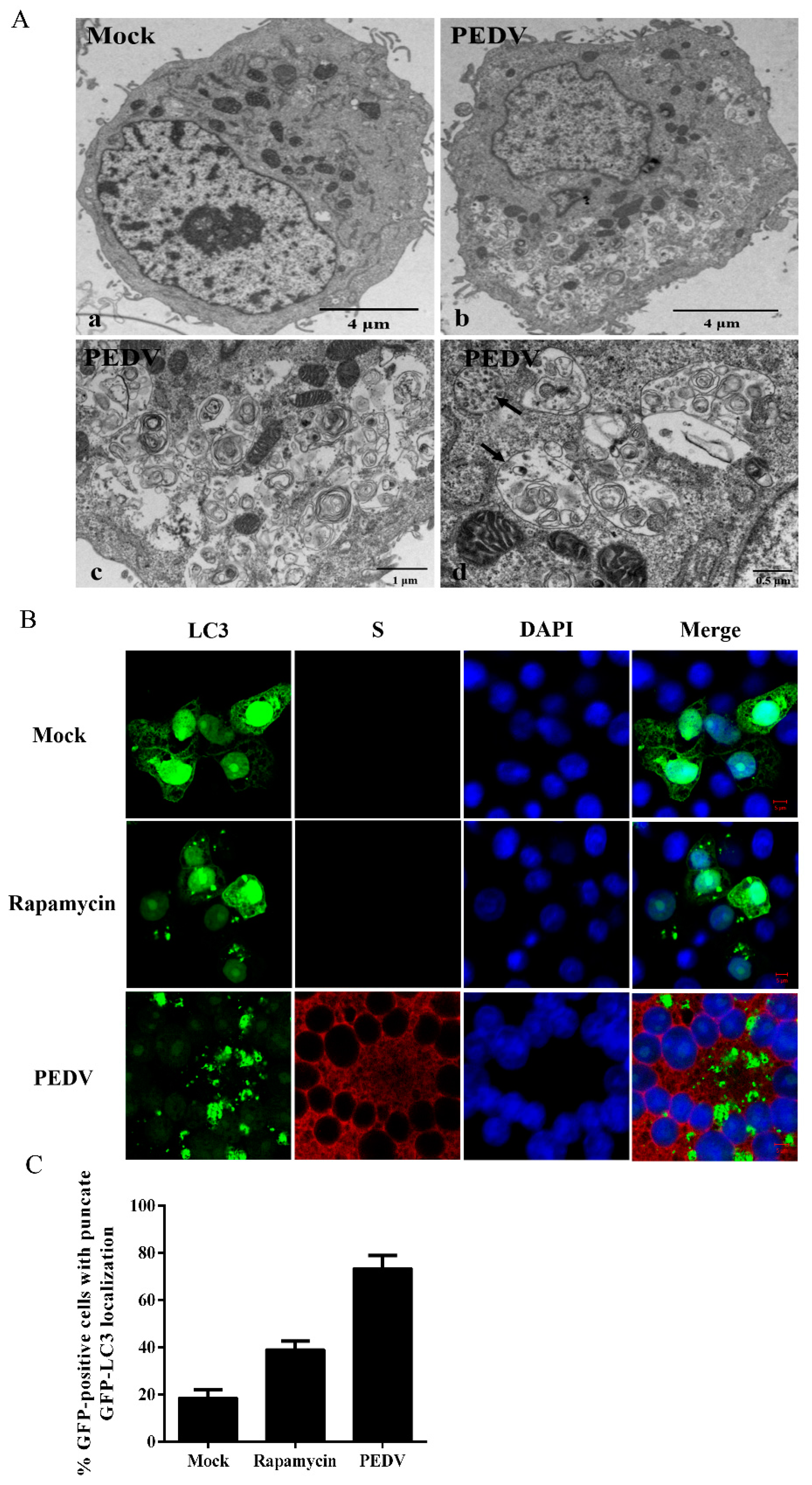
2.4. Transmission Electron Microscopy
2.5. Confocal Fluorescence Microscopy
2.6. Western Blot Analysis
2.7. RNA Interference
2.8. Cell Viability Assay
2.9. Statistical Analysis
3. Results
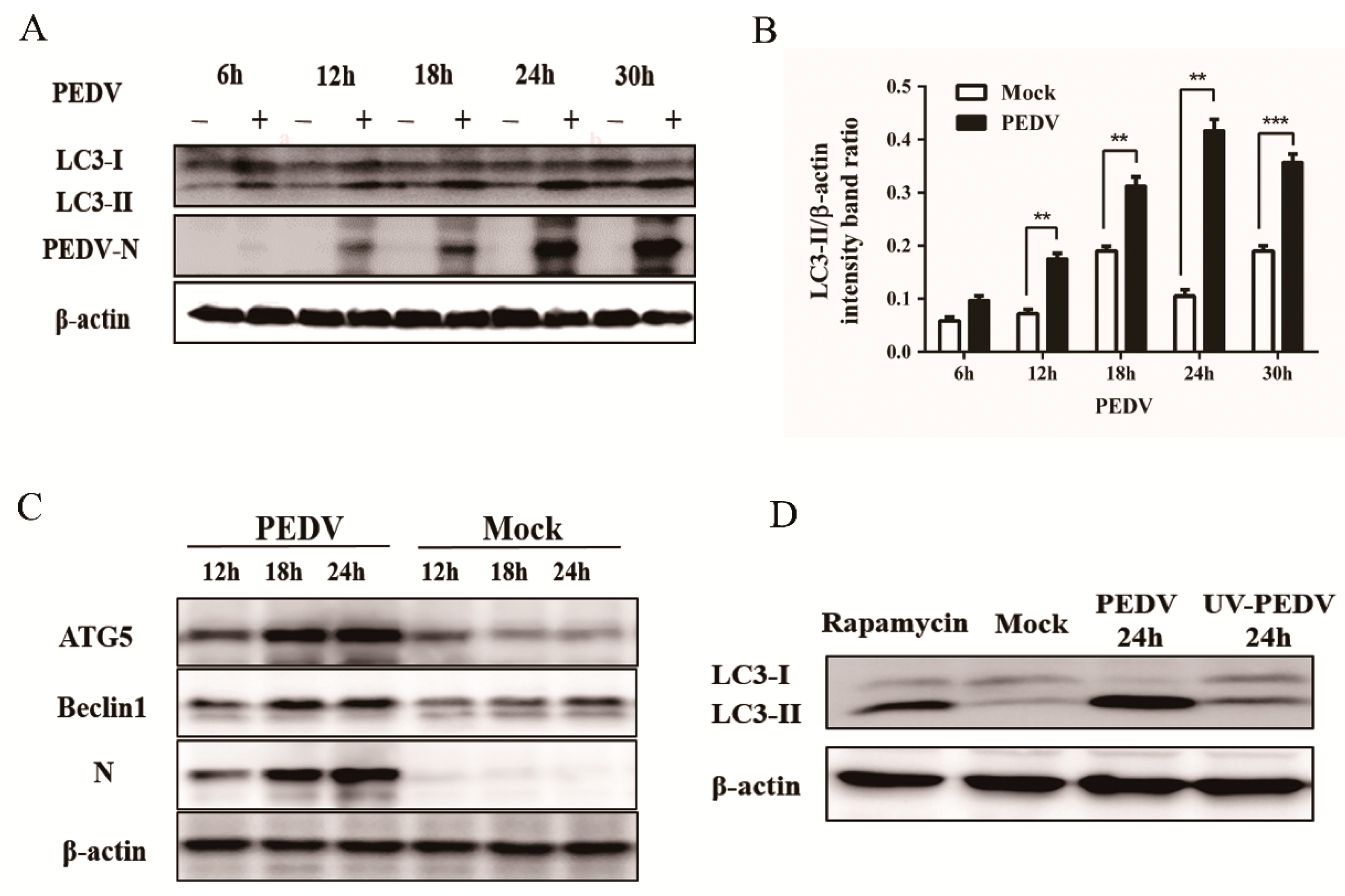
3.1. PEDV Infection Increases the Levels of Autophagy in Vero Cells
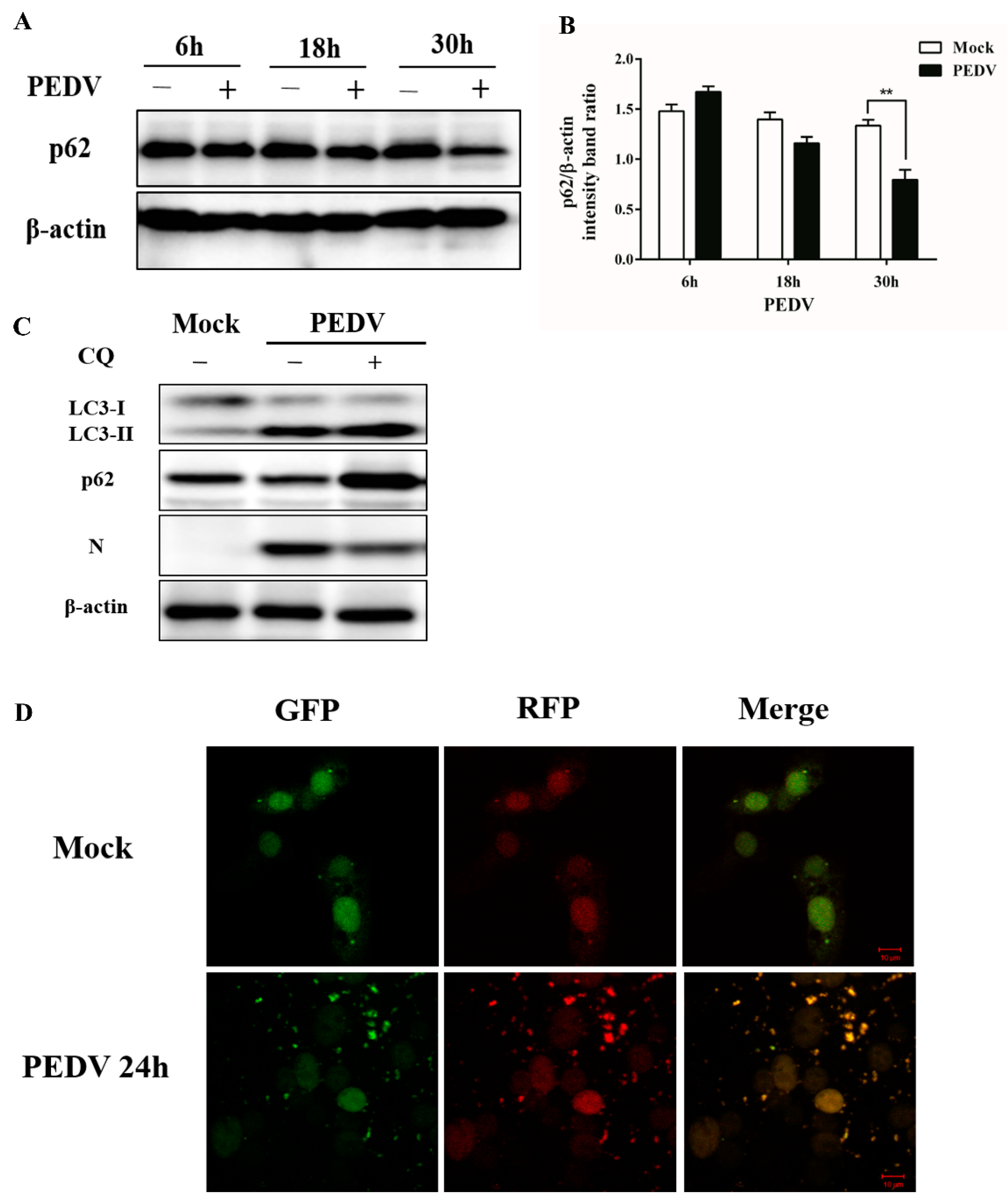
3.2. PEDV Infection Can Enhance Autophagy Flux
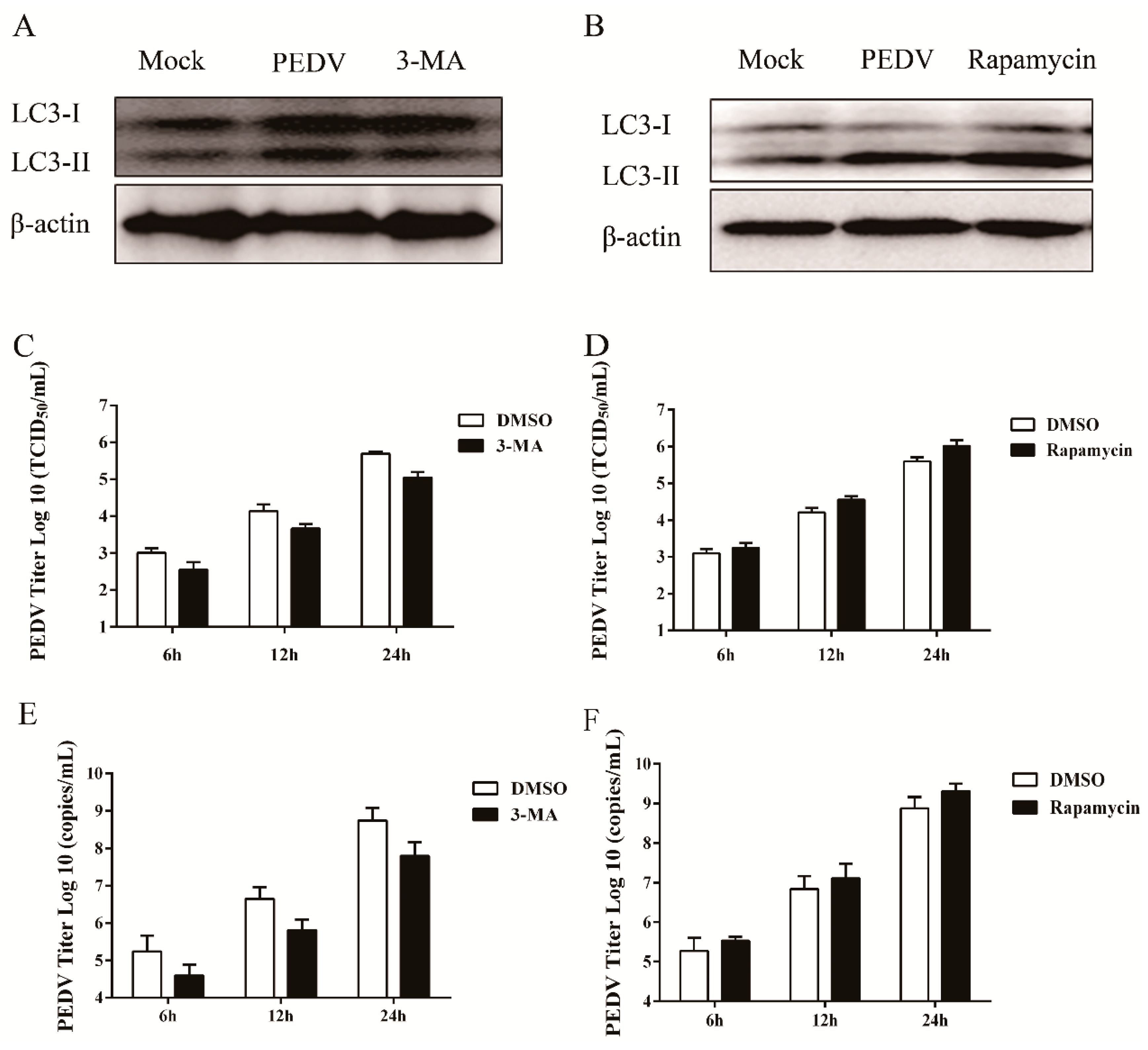
3.3. Pharmacological Inhibition of Autophagy Decreases Viral Yield
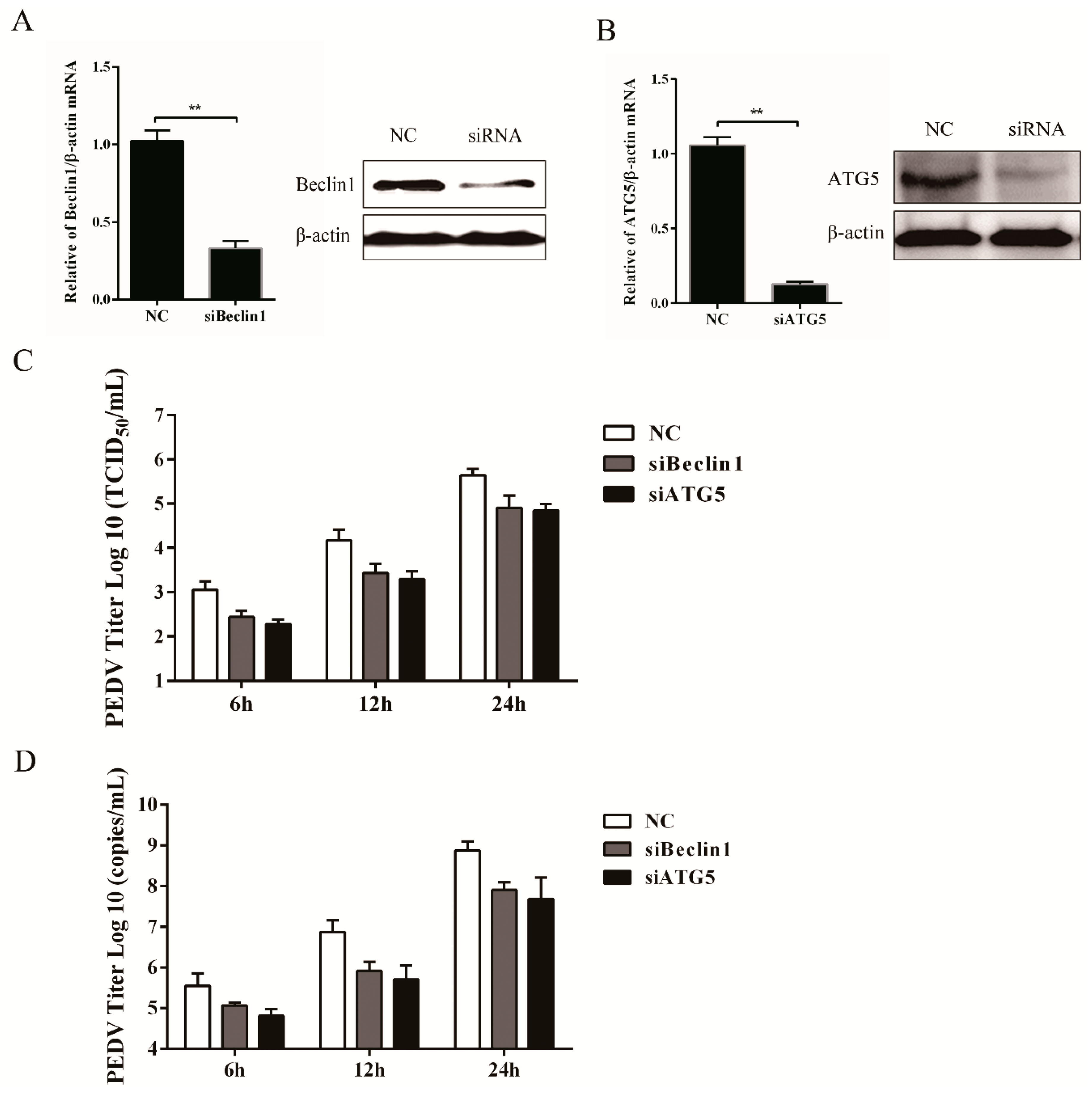
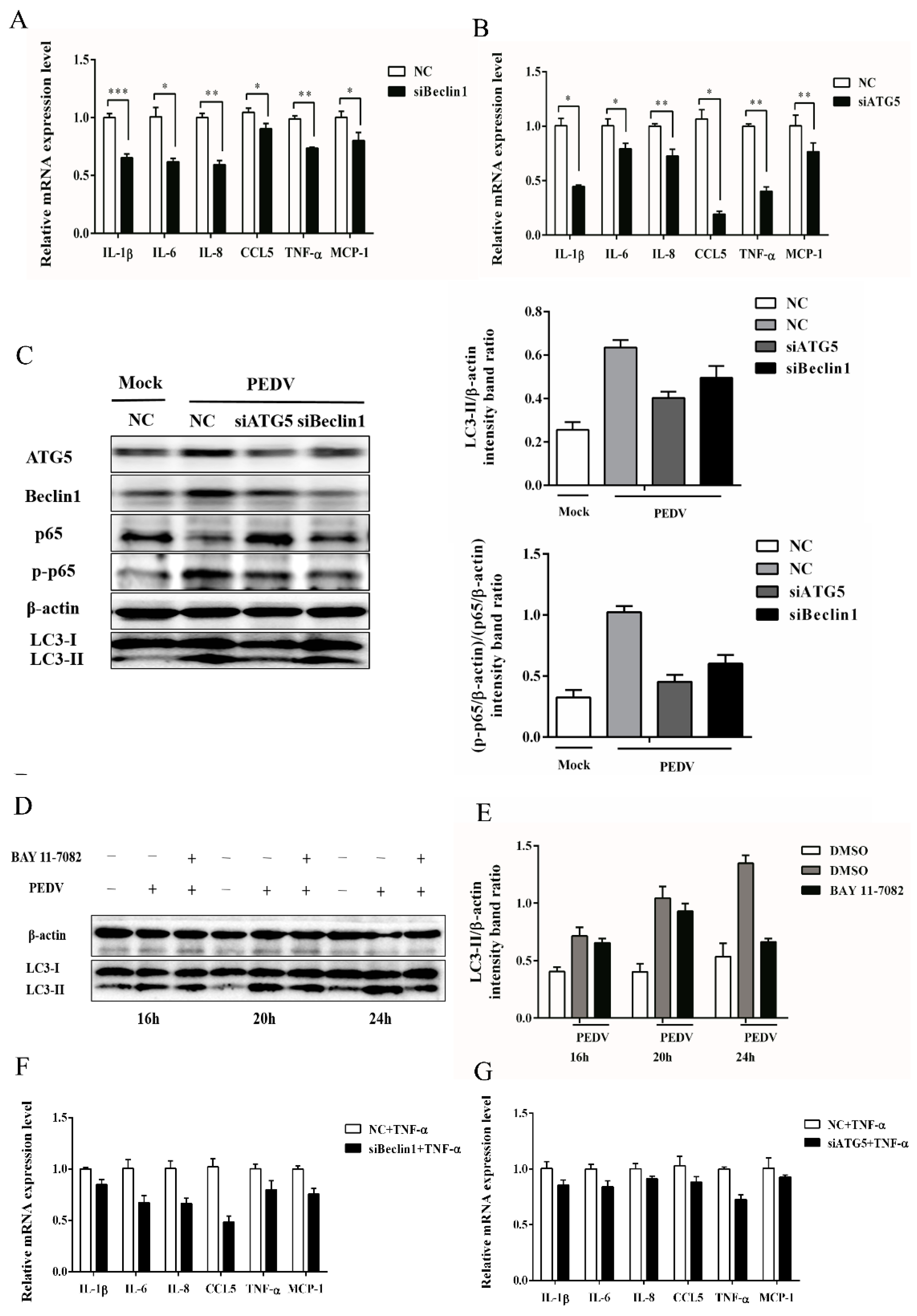
3.4. Silencing Endogenous Beclin1 or ATG5 Gene Reduces the PEDV Titer
3.5. Autophagy Has a Positive Correlation with NF-κB Signaling Pathway
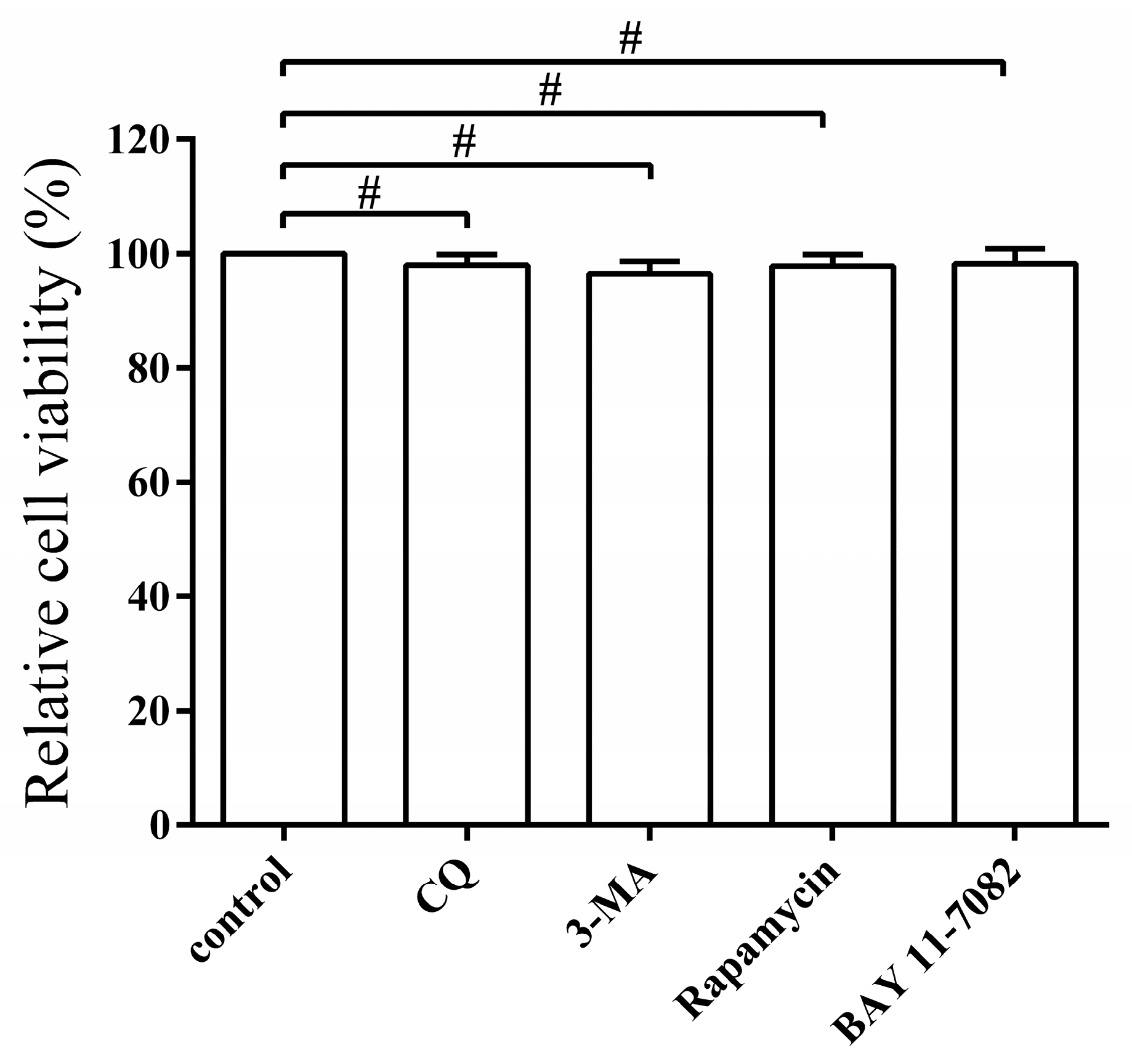
3.6. Pharmacological Regulation of Autophagy Does Not Affect Cell Viability
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, W.; Li, H.; Liu, Y.; Pan, Y.; Deng, F.; Song, Y.; Tang, X.; He, Q. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg. Infect. Dis. 2012, 18, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Crawford, K.; Lager, K.M.; Kulshreshtha, V.; Miller, L.C.; Faaberg, K.S. Status of vaccines for porcine epidemic diarrhea virus in the United States and Canada. Virus Res. 2016, 226, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T.; Kubota, T.; Ujike, M.; Yahara, Y.; Taguchi, F. Phylogenetic and antigenic characterization of newly isolated porcine epidemic diarrhea viruses in Japan. Virus Res. 2016, 222, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Dastjerdi, A.; Carr, J.; Ellis, R.J.; Steinbach, F.; Williamson, S. Porcine epidemic diarrhea virus among farmed pigs, ukraine. Emerg. Infect. Dis. 2015, 21, 2235–2237. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.; Gelfi, J.; Lambert, P.; Rasschaert, D.; Laude, H. Genome organization of porcine epidemic diarrhoea virus. Adv. Exp. Med. Biol. 1993, 342, 55–60. [Google Scholar] [PubMed]
- Song, D.; Park, B. Porcine epidemic diarrhoea virus: A comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes 2012, 44, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shi, K.; Yoo, D. Suppression of type I interferon production by porcine epidemic diarrhea virus and degradation of creb-binding protein by NSP1. Virology 2016, 489, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, S.; Jung, J.U. When autophagy meets viruses: A double-edged sword with functions in defense and offense. Semin. Munopathol. 2010, 32, 323–341. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, S.; Ding, N.; Meng, C.; Meng, S.; Zhang, S.; Zhan, Y.; Qiu, X.; Tan, L.; Chen, H.; et al. Autophagy benefits the replication of newcastle disease virus in chicken cells and tissues. J. Virol. 2014, 88, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Levine, B. Viruses and autophagy. Rev. Med. Virol. 2009, 19, 359–378. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Chisari, F.V. Viruses and the autophagy machinery. Cell Cycle 2010, 9, 1295–1307. [Google Scholar] [CrossRef] [PubMed]
- Chaumorcel, M.; Souquere, S.; Pierron, G.; Codogno, P.; Esclatine, A. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 2008, 4, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Jiang, W.; Virgin, H.W.T.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Zhao, M.; Ye, Z.; Gou, H.; Wang, J.; Yi, L.; Dong, X.; Liu, W.; Luo, Y.; Liao, M.; et al. Autophagy enhances the replication of classical swine fever virus in vitro. Autophagy 2014, 10, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.X.; Huang, L.; Wang, R.; Yu, Y.L.; Li, C.; Li, P.P.; Hu, X.C.; Hao, H.P.; Ishag, H.A.; Mao, X. Porcine reproductive and respiratory syndrome virus induces autophagy to promote virus replication. Autophagy 2012, 8, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Berkova, Z.; Crawford, S.E.; Trugnan, G.; Yoshimori, T.; Morris, A.P.; Estes, M.K. Rotavirus nsp4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J. Virol. 2006, 80, 6061–6071. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Shi, H.; Guo, D.; Chen, J.; Shi, D.; Zhu, Q.; Zhang, X.; Feng, L. Analysis of protein expression changes of the vero E6 cells infected with classic pedv strain CV777 by using quantitative proteomic technique. J. Virol. Methods 2015, 218, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Hu, H.; Chen, F.; Li, Z.; Ye, S.; Cheng, S.; Zhang, M.; He, Q. Itraq-based comparative proteomic analysis of vero cells infected with virulent and CV777 vaccine strain-like strains of porcine epidemic diarrhea virus. J. Proteom. 2016, 130, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Yan, W.; Gu, W.; He, Q. The ubiquitin-proteasome system is required for the early stages of porcine circovirus type 2 replication. Virology 2014, 456–457, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, C. Porcine epidemic diarrhea virus induces caspase-independent apoptosis through activation of mitochondrial apoptosis-inducing factor. Virology 2014, 460–461, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Cali, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses hijack the LC3-I-positive EDEmosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mo mMaas, A.M.; Snijder, E.J. Sars-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus NSP6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Fang, L.; Wang, D.; Wang, S.; Li, P.; Li, M.; Luo, R.; Chen, H.; Xiao, S. Induction of autophagy enhances porcine reproductive and respiratory syndrome virus replication. Virus Res. 2012, 163, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate i mMune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Zhang, Y.; Luo, Z.; Li, P.; Liu, L.; Wang, C.; Wang, H.; Li, H.; Ma, Y. Autophagy mediates avian influenza H5N1 pseudotyped particle-induced lung infla mMation through NF-kappab and p38 MAPK signaling pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L183–L195. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Ge, X.; Gao, Y.; Ren, Y.; Ren, X.; Li, G. Porcine epidemic diarrhea virus infection induces NF-kappab activation through the TLR2, TLR3 and TLR9 pathways in porcine intestinal epithelial cells. J. Gen. Virol. 2015, 96, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y.; Chen, S.S. Autophagy in hepatitis C virus-host interactions: Potential roles and therapeutic targets for liver-associated diseases. World J. Gastroenterol. 2014, 20, 5773–5793. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis c virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Cottam, E.M.; Stevenson-Leggett, P.; Wilkinson, J.A.; Harte, C.J.; Wileman, T.; Britton, P. Visualizing the autophagy pathway in avian cells and its application to studying infectious bronchitis virus. Autophagy 2013, 9, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.; Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in ma mMalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Richetta, C.; Gregoire, I.P.; Verlhac, P.; Azocar, O.; Baguet, J.; Flacher, M.; Tangy, F.; Rabourdin-Combe, C.; Faure, M. Sustained autophagy contributes to measles virus infectivity. PLoS Pathogens 2013, 9, e1003599. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Carpenter, J.E.; Jackson, W.; Grose, C. Autophagy and the effects of its inhibition on varicella-zoster virus glycoprotein biosynthesis and infectivity. J. Virol. 2014, 88, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.N.; Denison, M.R.; Virgin, H.W.T. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007, 3, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Petiot, A.; Ogier-Denis, E.; Blo mMaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Beclin 1 bridges autophagy, apoptosis and differentiation. Autophagy 2008, 4, 947–948. [Google Scholar] [CrossRef] [PubMed]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, M.L.; Piacentini, M. New insights on the role of apoptosis and autophagy in hiv pathogenesis. Apoptosis 2009, 14, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Xiao, Z.; Ma, X.; He, F.; Yao, H.; Liu, Z. Coxsackievirus b3 induces crosstalk between autophagy and apoptosis to benefit its release after replicating in autophagosomes through a mechanism involving caspase cleavage of autophagy-related proteins. Infect. Genet. Evol. 2014, 26, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza a virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Li, S.; Khan, F.A.; Zhang, S. Autophagy postpones apoptotic cell death in prrsv infection through bad-beclin1 interaction. Virulence 2016, 7, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, A.; Wang, J.; Zhang, S. Interplay of autophagy and apoptosis during prrsv infection of marc145 cell. Infect. Genet. Evol. 2016, 39, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Macian, F. Autophagy and the regulation of the I mMune response. Pharmacol. Res. 2012, 66, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in I mMunity and infla mMation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Papademetrio, D.L.; Lompardia, S.L.; Simunovich, T.; Costantino, S.; Mihalez, C.Y.; Cavaliere, V.; Alvarez, E. Inhibition of survival pathways MAPK and NF-kb triggers apoptosis in pancreatic ductal adenocarcinoma cells via suppression of autophagy. Target Oncol. 2016, 11, 183–195. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5’–3’) |
|---|---|
| PEDV-F | CGTACAGGTAAGTCAATTAC |
| PEDV-R | GATGAAGCATTGACTGAA |
| PEDV-probe-M | TTCGTCACAGTCGCCAAGG |
| ATG5-F | TTCACGCTATATCAGGAT |
| ATG5-R | ATCTCACTAATGTCTTCTTG |
| Beclin1-F | TGGCACAATCAATAACTTC |
| Beclin1-R | CAAGCAGCATTAATCTCAT |
| IL-6-F | TGTGAAAGCAGCAAAGAG |
| IL-6-R | AGTGTCCTCATTGAATCCA |
| IL-1β-F | GCGGCAACGAGGATGACTT |
| IL-1β-R | TGGCTACAACAACTGACACGG |
| IL-8-F | GGAACCATCTCGCTCTGTGTAA |
| IL-8-R | GGTCCACTCTCAATCACTCTCAG |
| CCL5-F | ACGCCTCGCTGTCATCCT |
| CCL5-R | GCACTTGCCACTGGTGTAGAA |
| TNF-α-F | CACCACGCTCTTCTGTCT |
| TNF-α-R | AGATGATCTGACTGCCTGAG |
| MCP-1-F | CTTCTGTGCCTGCTGCTCATA |
| MCP-1-R | ACTTGCTGCTGGTGATTCTTCT |
| GAPDH-F | ACATCATCCCTGCCTCTACTG |
| GAPDH-R | CCTGCTTCACCACCTTCTTG |
| β-actin-F | TTAGTTGCGTTACACCCTTTC |
| β-actin-R | ACCTTCACCGTTCCAGTT |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, X.; Zhang, M.; Zhang, X.; Tan, X.; Guo, H.; Zeng, W.; Yan, G.; Memon, A.M.; Li, Z.; Zhu, Y.; et al. Porcine Epidemic Diarrhea Virus Induces Autophagy to Benefit Its Replication. Viruses 2017, 9, 53. https://doi.org/10.3390/v9030053
Guo X, Zhang M, Zhang X, Tan X, Guo H, Zeng W, Yan G, Memon AM, Li Z, Zhu Y, et al. Porcine Epidemic Diarrhea Virus Induces Autophagy to Benefit Its Replication. Viruses. 2017; 9(3):53. https://doi.org/10.3390/v9030053
Chicago/Turabian StyleGuo, Xiaozhen, Mengjia Zhang, Xiaoqian Zhang, Xin Tan, Hengke Guo, Wei Zeng, Guokai Yan, Atta Muhammad Memon, Zhonghua Li, Yinxing Zhu, and et al. 2017. "Porcine Epidemic Diarrhea Virus Induces Autophagy to Benefit Its Replication" Viruses 9, no. 3: 53. https://doi.org/10.3390/v9030053
APA StyleGuo, X., Zhang, M., Zhang, X., Tan, X., Guo, H., Zeng, W., Yan, G., Memon, A. M., Li, Z., Zhu, Y., Zhang, B., Ku, X., Wu, M., Fan, S., & He, Q. (2017). Porcine Epidemic Diarrhea Virus Induces Autophagy to Benefit Its Replication. Viruses, 9(3), 53. https://doi.org/10.3390/v9030053