
Single-Round Infectious Particle Antiviral Screening Assays for the Japanese Encephalitis Virus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Cells and Virus
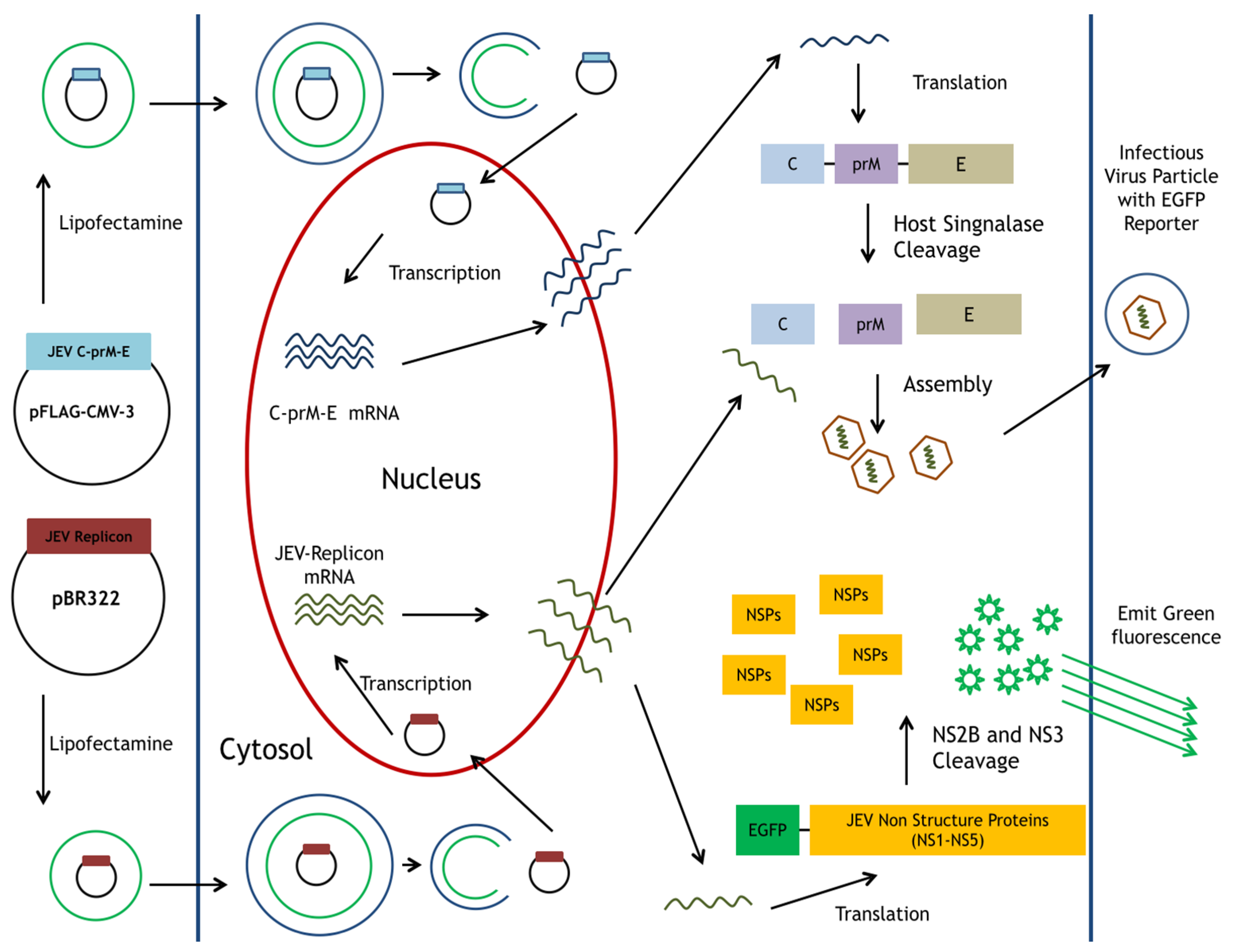
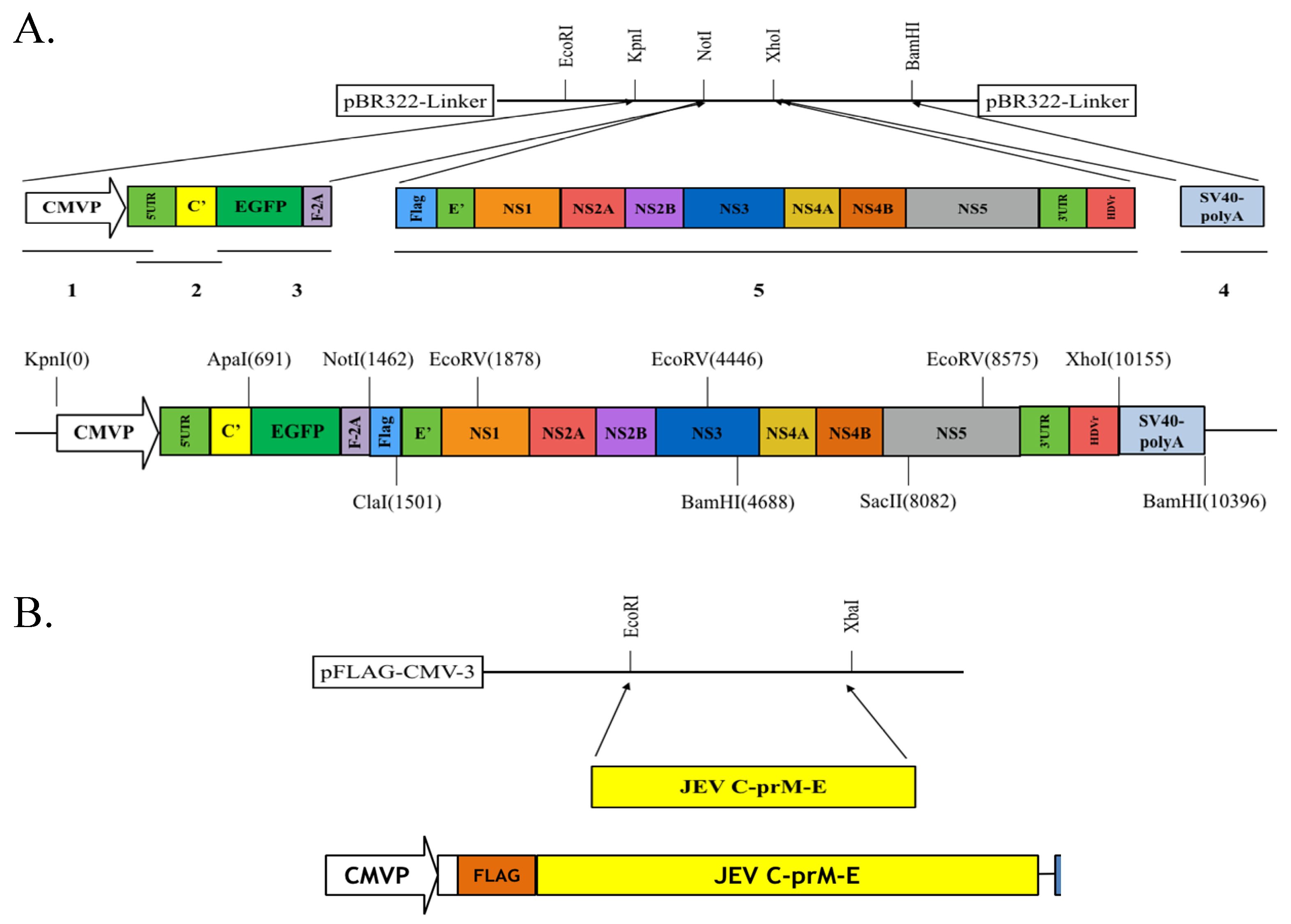
2.2. Preparation of pBR322-Based JEV-EGFP Replicon and pFlag-CMV3-CprME
2.3. Functional Analysis of JEV-EGFP Replicon Using RT- PCR and Immunofluorescent Staining
2.4. Generation of a TE671 Cell Line Expressing C, prM/M, and E Proteins for the Production of JEV-EGFP SRIPs
2.5. Infective Activity Assay of JEV-EGFP SRIPs Using Flow Cytometry
2.6. Synthesis of Compound MJ-47
2.7. Cytotoxicity of Compound MJ-47 with a MTT Assay
2.8. Inhibitory Assays of Cytopathic Effect and Replicon-Driven Replication by MJ-47
2.9. A Combination Assay of JEV-EGFP SRIP and Flow Cytometry for IC50
2.10. Plaque Reduction Assay for IC50
2.11. Virucidal and Attachment Assays
2.12. Statistical Analysis
3. Result
3.1. Functional Analysis of pBR322 Plasmid-Based JEV Replicon
3.2. Production and Infectivity of JEV-EGFP SRIPs
3.3. Evaluation of the Antiviral Screening Platform Using JEV-EGFP SRIPs and Flow Cytometry
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weaver, S.C.; Reisen, W.K. Present and future arboviral threats. Antivir. Res. 2010, 85, 328–345. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Diamond, M.S. Zika virus: New clinical syndromes and its emergence in the Western Hemisphere. J. Virol. 2016, 90, 4864–4875. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Ni, H.; Beasley, D.W.; Ekkelenkamp, M.; Cardosa, M.J.; Barrett, A.D. Origin and evolution of Japanese Encephalitis virus in Southeast Asia. J. Virol. 2003, 77, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Markoff, L.; Falgoutm, B.; Chang, A. A conserved internal hydrophobic domain mediates the stable membrane integration of the dengue viruscapsid protein. Virology 1997, 233, 105–117. [Google Scholar] [CrossRef] [PubMed]
- De Wispelaere, M.; Khou, C.; Frenkiel, M.P.; Desprès, P.; Pardigon, N. A single amino acid substitution in the M protein attenuates Japanese Encephalitis virus in mammalian hosts. J. Virol. 2015, 90, 2676–2689. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, H.; Li, Z.; Wang, W.; Lin, H.; Liu, L.; Ni, Q.; Liu, X.; Zeng, X.; Wu, Y.; et al. Envelope protein mutations L107F and E138K are important for neurovirulence attenuation for Japanese Encephalitis virus SA14-14-2 strain. Viruses 2017. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Xu, Y.P.; Zhang, Y.; Li, X.F.; Wang, H.J.; Liu, Z.Y.; Li, S.H.; Liu, L.; Zhao, H.; Nian, Q.G.; et al. Genotype-specific neutralization determinants in envelope protein: Implications for the improvement of Japanese encephalitis vaccine. J. Gen. Virol. 2015, 96, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 1996, 220, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. trans-Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J. Virol. 1997, 71, 9608–9617. [Google Scholar] [PubMed]
- Xie, X.; Gayen, S.; Kang, C.; Yuan, Z.; Shi, P.Y. Membrane topology and function of dengue virus NS2A protein. J. Virol. 2013, 87, 4609–4622. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Unno, H.; Mori, Y.; Tani, H.; Moriishi, K.; Takamizawa, A.; Agoh, M.; Tsukihara, T.; Matsuura, Y. Crystal structure of the catalytic domain of Japanese Encephalitis virus NS3 helicase/nucleoside triphosphatase at a resolution of 1.8 A. Virology 2008, 373, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Huang, H.D.; Shiu, S.Y.; Chen, W.J.; Tsai, M.H.; Huang, S.H.; Wan, L.; Lin, Y.J. Functional determinants of NS2B for activation of Japanese Encephalitis virus NS3 protease. Virus Res. 2007, 127, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Bühler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K- regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Jordán, J.L.; Laurent-Rolle, M.; Ashour, J.; Martínez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; García-Sastre, A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, K.; Igarashi, M.; Ito, K.; Kariwa, H.; Holbrook, M.R.; Takashima, I. Construction of an infectious cDNA clone for Omsk hemorrhagic fever virus, and characterization of mutations in NS2A and NS5. Virus Res. 2011, 155, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Unni, S.K.; Růžek, D.; Chhatbar, C.; Mishra, R.; Johri, M.K.; Singh, S.K. Japanese Encephalitis virus: From genome to infectome. Microbes Infect. 2011, 13, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Hombach, J.; Solomon, T.; Kurane, I.; Jacobson, J.; Wood, D. Report on a WHO consultation on immunological endpoints for evaluation of new Japanese encephalitis vaccines, WHO, Geneva, 2–3 September, 2004. Vaccine 2005, 23, 5205–5211. [Google Scholar] [CrossRef] [PubMed]
- Nga, P.T.; del Carmen, P.M.; Cuong, V.D.; Ma, S.P.; Hasebe, F.; Inoue, S.; Makino, Y.; Takagi, M.; Nam, V.S.; Morita, K. Shift in Japanese Encephalitis virus (JEV) genotype circulating in northern Vietnam: implications for frequent introductions of JEV from southeast Asia to east Asia. J. Gen. Virol. 2004, 85, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Fu, S.; Gao, X.; Li, M.; Cui, S.; Li, X.; Cao, Y.; Lei, W.; Lu, Z.; He, Y.; et al. Low protective efficacy of the current Japanese encephalitis vaccine against the emerging genotype 5 Japanese Encephalitis virus. PLoS Negl. Trop. Dis. 2016, 10, e0004686. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Yi, M.; Li, K.; Lemon, S.M. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J. Virol. 2002, 76, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Almazán, F.; Sola, I.; Zuñiga, S.; Marquez-Jurado, S.; Morales, L.; Becares, M.; Enjuanes, L. Coronavirus reverse genetic systems: Infectious clones and replicons. Virus Res. 2014, 189, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Leardkamolkarn, V.; Sirigulpanit, W.; Chotiwan, N.; Kumkate, S.; Huang, C.Y.-H. Development of Dengue type-2 virus replicons expressing GFP reporter gene in study of viral RNA replication. Virus Res. 2012, 163, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Fayzulin, R.; Scholle, F.; Petrakova, O.; Frolov, I.; Mason, P.W. Evaluation of replicative capacity and genetic stability of West Nile virus replicons using highly efficient packaging cell lines. Virology 2006, 351, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Murata, R.; Akiyama, M.; Takashima, I.; Kariwa, H.; Watanabe, T.; Kurane, I.; Maeda, J. A PCR-based protocol for generating West Nile virus replicons. J. Virol. Methods 2008, 148, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Sumiyoshi, H.; Hoke, C.H.; Trent, D.W. Infectious Japanese Encephalitis virus RNA can be synthesized from in vitro-ligated cDNA templates. J. Virol. 1992, 66, 5425–5431. [Google Scholar] [PubMed]
- Li, S.H.; Li, X.F.; Zhao, H.; Deng, Y.Q.; Yu, X.D.; Zhu, S.Y.; Jiang, T.; Ye, Q.; Qin, E.D.; Qin, C.F. Development and characterization of the replicon system of Japanese encephalitis live vaccine virus SA14-14-2. Virology 2013, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Mohr, H.; Mohr, C.A.; Schneider, M.R.; Scrivano, L.; Adler, B.; Kraner-Schreiber, S.; Schnieke, A.; Dahlhoff, M.; Wolf, E.; Koszinowski, U.H.; et al. Cytomegalovirus replicon-based regulation of gene expression in vitro and in vivo. PLoS Pathog. 2012, 8, e1002728. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, S.; Yang, P.; Wang, C.; Du, Y.; Yu, W.; Sun, Z. Replicon-based Japanese Encephalitis virus vaccines elicit immune response in mice. J. Virol. Methods 2012, 179, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Tsai, M.H.; Hu, H.S.; Pu, S.Y.; Wu, R.H.; Wu, S.H.; Lin, H.M.; Song, J.S.; Chao, Y.S.; Yueh, A. Characterization of an efficient dengue virus replicon for development of assays of discovery of small molecules against dengue virus. Antivir. Res. 2013, 98, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Varnavski, A.N.; Khromykh, A.A. Noncytopathic flavivirus replicon RNA- based system for expression and delivery of heterologous genes. Virology 1999, 255, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.C.; Liu, W.J.; Anraku, I.; Clark, D.C.; Pollitt, C.C.; Suhrbier, A.; Hall, R.A.; Khromykh, A.A. Single-round infectious particles enhance immunogenicity of a DNA vaccine against West Nile virus. Nat. Biotechnol. 2008, 26, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, K.; Ikawa, A.; Chiba, Y.; Omori, Y.; Maeda, J.; Murata, R.; Kariwa, H.; Takashima, I. Establishment of a neutralization test involving reporter gene-expressing virus-like particles of tick-borne Encephalitis virus. J. Virol. Methods 2009, 161, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Ishikawa, T.; Konishi, E.; Matsuda, M.; Watashi, K.; Aizaki, H.; Takasaki, T.; Wakita, T. Production of single-round infectious chimeric flaviviruses with DNA-based Japanese Encephalitis virus replicon. J. Gen. Virol. 2014, 95, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, A.; Suzuki, R.; Konishi, E. Evaluation of single-round infectious, chimeric dengue type 1 virus as an antigen for dengue functional antibody assays. Vaccine 2014, 32, 4289–4295. [Google Scholar] [CrossRef] [PubMed]
- Roby, J.A.; Bielefeldt-Ohmann, H.; Prow, N.A.; Chang, D.C.; Hall, R.A.; Khromykh, A.A. Increased expression of capsid protein in trans enhances production of single-round infectious particles by West Nile virus DNA vaccine candidate. J. Gen. Virol. 2014, 95, 2176–2191. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.T.; Liao, J.T.; Yen, L.C.; Chang, Y.K.; Lin, Y.L.; Liao, C.L. Japanese Encephalitis virus replicon-based vaccine expressing enterovirus-71 epitope confers dual protection from lethal challenges. J. Biomed. Sci. 2015, 22, 74. [Google Scholar] [CrossRef] [PubMed]
- Hour, M.J.; Huang, L.J.; Kuo, S.C.; Xia, Y.; Bastow, K.; Nakanishi, Y.; Hamel, E.; Lee, K.H. 6-Alkylamino- and 2,3-dihydro-3′-methoxy-2-phenyl-4-quinazolinones and related compounds: Their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J. Med. Chem. 2000, 43, 4479–4487. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Abe, M.; Masuda, M. Construction of an infectious molecular clone of Japanese Encephalitis virus genotype V and its derivative subgenomic replicon capable of expressing a foreign gene. Virus Res. 2015, 195, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Tong, W.; Liu, F.; Liang, C.; Gao, F.; Li, G.; Tong, G.; Zheng, H. Genetic instability of Japanese Encephalitis virus cDNA clones propagated in Escherichia coli. Virus Genes 2016, 52, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Pu, S.Y.; Wu, R.H.; Yang, C.C.; Jao, T.M.; Tsai, M.H.; Wang, J.C.; Lin, H.M.; Chao, Y.S.; Yueh, A. Successful propagation of flavivirus infectious cDNAs by a novel method to reduce the cryptic bacterial promoter activity of virus genomes. J. Virol. 2011, 85, 2927–2941. [Google Scholar] [CrossRef] [PubMed]
- Bollati, M.; Alvarez, K.; Assenberg, R.; Baronti, C.; Canard, B.; Cook, S.; Coutard, B.; Decroly, E.; de Lamballerie, X.; Gould, E.A.; et al. Structure and functionality in flavivirus NS-proteins: Perspectives for drug design. Antiviral Res. 2010, 87, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Gong, P. Crystal structure of the full-length Japanese Encephalitis virus NS5 reveals a conserved methyltransferase-polymerase interface. PLoS Pathog. 2013, 9, e1003549. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Nukui, Y.; Tajima, S.; Nerome, R.; Kato, F.; Watanabe, H.; Takasaki, T.; Kurane, I. An amino acid substitution (V3I) in the Japanese Encephalitis virus NS4A protein increases its virulence in mice, but not its growth rate in vitro. J. Gen. Virol. 2011, 92, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Li, X.F.; Ye, H.Q.; Deng, C.L.; Ye, Q.; Shan, C.; Shang, B.D.; Xu, L.L.; Li, S.H.; Cao, S.B.; et al. Recovery of a chemically synthesized Japanese Encephalitis virus reveals two critical adaptive mutations in NS2B and NS4A. J. Gen. Virol. 2014, 95, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.K.; Scherbarth, A.; Sprengel, R.; Engelhardt, J.; Theer, P.; Giese, G. Fluorescent-protein stabilization and high-resolution imaging of cleared, intact mouse brains. PLoS ONE 2015, 10, e0124650. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, D.G.; Andrei, G.; Schols, D.; Snoeck, R.; Grabkowska-Drużyc, M. New isoxazolidine-conjugates of quinazolinones-synthesis, antiviral and cytostatic activity. Molecules 2016, 21, 959. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, W.; Jiang, L.; Wan, S.; Zhang, L.; Yu, R.; Jiang, T. 2-Pyridinyl-4(3H)-quinazolinone: A scaffold for anti-influenza a virus compounds. Chem. Biol. Drug Des. 2015, 86, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.T.; Butcher, J.W.; Nguyen, K.T.; McIntyre, C.J.; Romano, J.J.; Gilbert, K.F.; Bush, K.J.; Liverton, N.J.; Holloway, M.K.; Harper, S.; et al. P2-quinazolinones and bis-macrocycles as new templates for next-generation hepatitis C virus NS3/4a protease inhibitors: Discovery of MK-2748 and MK-6325. Chem. Med. Chem. 2015, 10, 727–735. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.-Y.; Hour, M.-J.; Wang, C.-Y.; Huang, S.-H.; Mu, W.-X.; Chang, Y.-C.; Lin, C.-W. Single-Round Infectious Particle Antiviral Screening Assays for the Japanese Encephalitis Virus. Viruses 2017, 9, 76. https://doi.org/10.3390/v9040076
Lu C-Y, Hour M-J, Wang C-Y, Huang S-H, Mu W-X, Chang Y-C, Lin C-W. Single-Round Infectious Particle Antiviral Screening Assays for the Japanese Encephalitis Virus. Viruses. 2017; 9(4):76. https://doi.org/10.3390/v9040076
Chicago/Turabian StyleLu, Chien-Yi, Mann-Jen Hour, Ching-Ying Wang, Su-Hua Huang, Wen-Xiang Mu, Yu-Chun Chang, and Cheng-Wen Lin. 2017. "Single-Round Infectious Particle Antiviral Screening Assays for the Japanese Encephalitis Virus" Viruses 9, no. 4: 76. https://doi.org/10.3390/v9040076
APA StyleLu, C. -Y., Hour, M. -J., Wang, C. -Y., Huang, S. -H., Mu, W. -X., Chang, Y. -C., & Lin, C. -W. (2017). Single-Round Infectious Particle Antiviral Screening Assays for the Japanese Encephalitis Virus. Viruses, 9(4), 76. https://doi.org/10.3390/v9040076