
Differentiation and Structure in Sulfolobus islandicus Rod-Shaped Virus Populations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Environmental Sampling
2.2. Enrichment Cultures of Environmental Samples
2.3. Viral Isolation and Purification
2.4. Virus Quantification and Cross-Infection Assays
2.5. DNA Extraction
2.6. Genome Sequencing and Assembly
2.7. Comparative Genomics and Phylogenetic Analysis of SIRVs
2.8. Phylogenetic Analyses of S. Islandicus Strains
2.9. Host Variation
2.10. Analysis of CRISPR Spacer Matches
3. Results
3.1. Sampling
3.2. SIRV Isolation and Structure
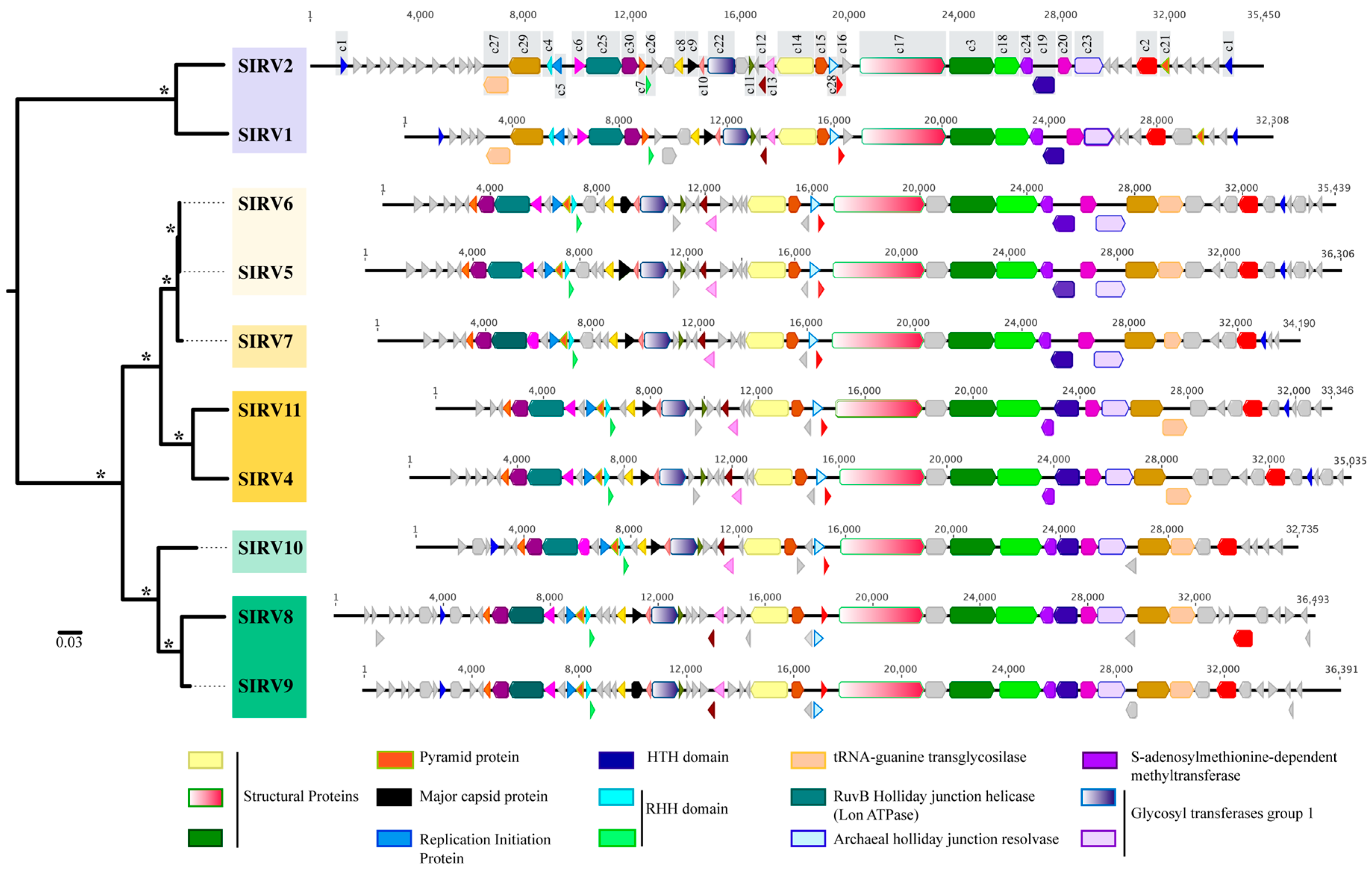
3.3. Comparative Genomics of Sulfolobus Islandicus Rod-Shaped Core Genome
3.3.1. Core Genome
3.3.2. Variable Genome
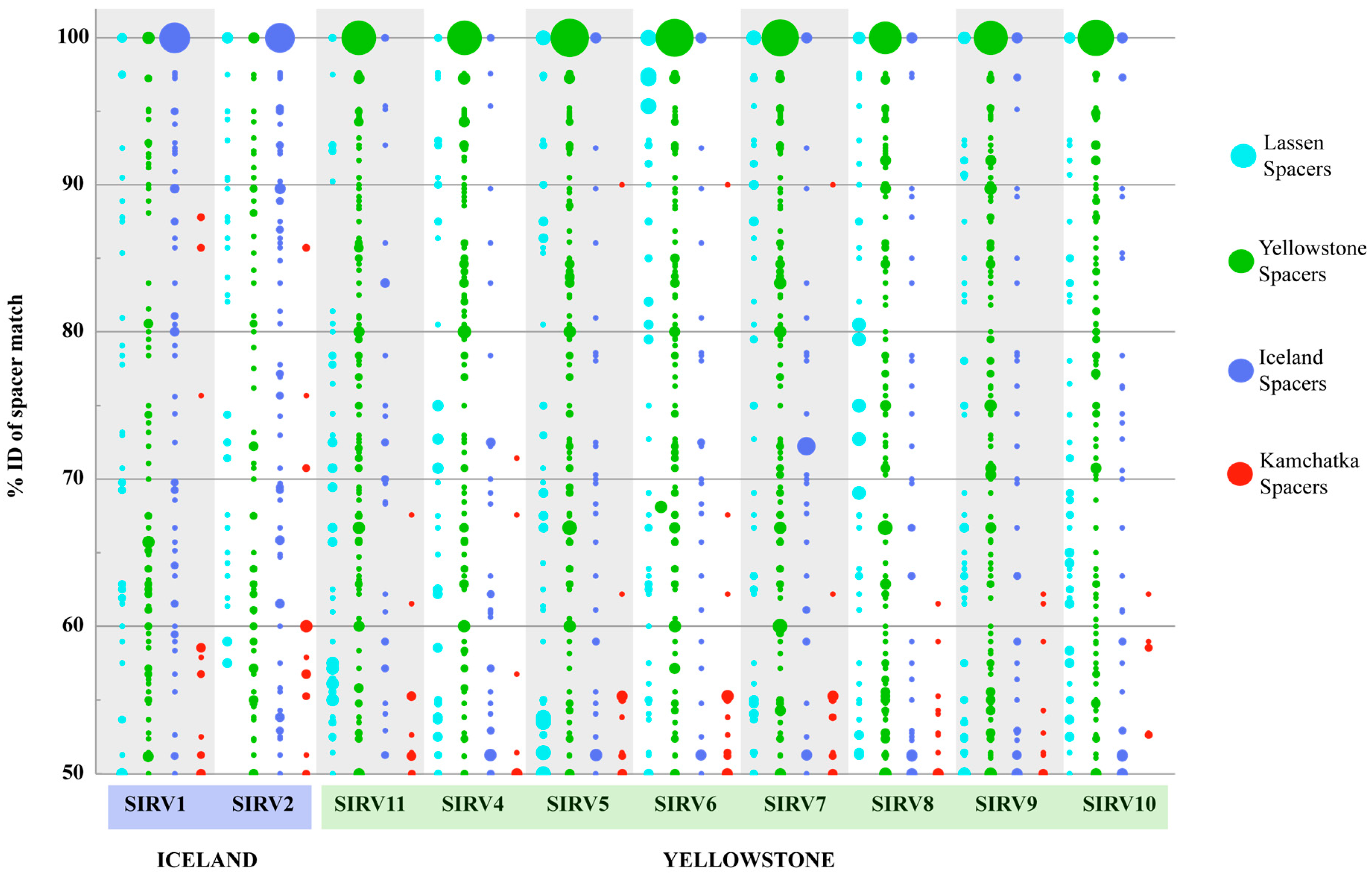
3.4. Host-Virus Interactions Recorded in the S. Islandicus CRISPR Repeat Spacer Arrays
3.5. Host Range of SIRVs
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Valera, F.; Mizuno, C.M.; Ghai, R. Tales from a thousand and one phages. Bacteriophage 2014, 4, e28265-1–e28265-6. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Thurber, R.V. Viruses manipulate the marine environment. Nature 2009, 459, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Prangishvili, D. The Wonderful World of Archaeal Viruses. Annu. Rev. Microbiol. 2013, 67, 565–585. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, A.C.; Wiedenheft, B.; Douglas, T.; Young, M. Hot crenarchaeal viruses reveal deep evolutionary connections. Nat. Rev. Microbiol. 2006, 4, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Prangishvili, D.; Forterre, P.; Garrett, R.A. Viruses of the Archaea: A unifying view. Nat. Rev. Microbiol. 2006, 4, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H.; Barnett, L.; Brenner, S.; Watts-Tobin, R.J. General nature of the genetic code for proteins. Nature 1961, 192, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Hershey, A.D.; Chase, M. Independent Functions of Viral Protein and Nucleic Acid in Growth of Bacteriophage. J. Gen. Physiol. 1952, 36, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Sullivan, M.B. Rising to the challenge: Accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol. 2015, 13, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Valera, F.; Martin-Cuadrado, A.-B.; Rodriguez-Brito, B.; Pasic, L.; Thingstad, T.F.; Rohwer, F.; Mira, A. Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 2009, 7, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Prangishvili, D.; Lindell, D. Roles of viruses in the environment. Environ. Microbiol. 2009, 11, 2771–2774. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Wirth, J.F.; Mazurie, A.; Young, M.J. Viral assemblage composition in Yellowstone acidic hot springs assessed by network analysis. ISME J. 2015, 9, 2162–2177. [Google Scholar] [CrossRef] [PubMed]
- Pina, M.; Bize, A.; Forterre, P.; Prangishvili, D. The archeoviruses. FEMS Microbiol. Rev. 2011, 35, 1035–1054. [Google Scholar] [CrossRef] [PubMed]
- Quax, T.E.F.; Krupovič, M.; Lucas, S.; Forterre, P.; Prangishvili, D. The Sulfolobus rod-shaped virus 2 encodes a prominent structural component of the unique virion release system in Archaea. Virology 2010, 404, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Rachel, R.; Bettstetter, M.; Hedlund, B.P.; Häring, M.; Kessler, A.; Stetter, K.O.; Prangishvili, D. Remarkable morphological diversity of viruses and virus-like particles in hot terrestrial environments. Arch. Virol. 2002, 147, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Quax, T.E.F.; Lucas, S.; Reimann, J.; Pehau-Arnaudet, G.; Prevost, M.-C.; Forterre, P.; Albers, S.-V.; Prangishvili, D. Simple and elegant design of a virion egress structure in Archaea. Proc. Natl. Acad. Sci. USA 2011, 108, 3354–3359. [Google Scholar] [CrossRef] [PubMed]
- Brumfield, S.K.; Ortmann, A.C.; Ruigrok, V.; Suci, P.; Douglas, T.; Young, M.J. Particle assembly and ultrastructural features associated with replication of the lytic archaeal virus Sulfolobus turreted icosahedral virus. J. Virol. 2009, 83, 5964–5970. [Google Scholar] [CrossRef] [PubMed]
- Prangishvili, D.; Koonin, E.V.; Krupovic, M. Genomics and biology of Rudiviruses, a model for the study of virus-host interactions in Archaea. Biochem. Soc. Trans. 2013, 41, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Prangishvili, D.; Arnold, H.P.; Gotz, D.; Ziese, U.; Holz, I.; Kristjansson, J.K.; Zillig, W. A novel virus family, the Rudiviridae: Structure, virus-host interactions and genome variability of the Sulfolobus viruses SIRV1 and SIRV2. Genetics 1999, 152, 1387–1396. [Google Scholar] [PubMed]
- Peng, X.; Blum, H.; She, Q.; Mallok, S.; Brügger, K.; Garrett, R.A.; Zillig, W.; Prangishvili, D. Sequences and Replication of Genomes of the Archaeal Rudiviruses SIRV1 and SIRV2: Relationships to the Archaeal Lipothrixvirus SIFV and Some Eukaryal Viruses. Virology 2001, 291, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Oke, M.; Kerou, M.; Liu, H.; Peng, X.; Garrett, R.A.; Prangishvili, D.; Naismith, J.H.; White, M.F. A Dimeric Rep Protein Initiates Replication of a Linear Archaeal Virus Genome: Implications for the Rep Mechanism and Viral Replication. J. Virol. 2011, 85, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Guillière, F.; Peixeiro, N.; Kessler, A.; Raynal, B.; Desnoues, N.; Keller, J.; Delepierre, M.; Prangishvili, D.; Sezonov, G.; Guijarro, J.I. Structure, function, and targets of the transcriptional regulator SvtR from the hyperthermophilic archaeal virus SIRV1. J. Biol. Chem. 2009, 284, 22222–22237. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kragelund, B.B.; White, M.F.; Peng, X. Functional Characterization of a Conserved Archaeal Viral Operon Revealing Single-Stranded DNA Binding, Annealing and Nuclease Activities. J. Mol. Biol. 2015, 427, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; He, F.; Bhoobalan-Chitty, Y.; Martinez-Alvarez, L.; Guo, Y.; Peng, X. Unveiling Cell Surface and Type IV Secretion Proteins Responsible for Archaeal Rudivirus Entry. J. Virol. 2014, 88, 10264–10268. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, F.; Yu, X.; Rensen, E.; Krupovic, M.; Prangishvili, D.; Egelman, E.H. A virus that infects a hyperthermophile encapsidates A-form DNA. Science 2015, 348, 914–917. [Google Scholar] [CrossRef] [PubMed]
- Daum, B.; Quax, T.E.F.; Sachse, M.; Mills, D.J.; Reimann, J.; Yildiz, Ö.; Häder, S.; Saveanu, C.; Forterre, P.; Albers, S.-V.; Kühlbrandt, W.; Prangishvili, D. Self-assembly of the general membrane-remodeling protein PVAP into sevenfold virus-associated pyramids. Proc. Natl. Acad. Sci. USA 2014, 111, 3829–3834. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Bautista, M.A.; Zhang, C.; Whitaker, R.J. Virus-Induced Dormancy in the Archaeon Sulfolobus islandicus. mBio 2015, 6, e02565-14. [Google Scholar] [CrossRef] [PubMed]
- Held, N.L.; Whitaker, R.J. Viral biogeography revealed by signatures in Sulfolobus islandicus genomes. Environ. Microbiol. 2009, 11, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.C.; Wiedenheft, B.; Lavin, M.; Roberto, F.F.; Spuhler, J.; Ortmann, A.C.; Douglas, T.; Young, M. Virus movement maintains local virus population diversity. Proc. Natl. Acad. Sci. USA 2007, 104, 19102–19107. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Gudbergsdóttir, S.R.; Rike, A.G.; Lin, L.; Zhang, Q.; Contursi, P.; Moracci, M.; Kristjansson, J.K.; Bolduc, B.; Gavrilov, S.; et al. Comparative Metagenomics of Eight Geographically Remote Terrestrial Hot Springs. Microb. Ecol. 2015, 70, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.J.; Grogan, D.W.; Taylor, J.W. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 2003, 301, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Schleper, C.; Kubo, K.; Zillig, W. The particle SSV1 from the extremely thermophilic archaeon Sulfolobus is a virus: Demonstration of infectivity and of transfection with viral DNA. Proc. Natl. Acad. Sci. USA 1992, 89, 7645–7649. [Google Scholar] [CrossRef] [PubMed]
- Fulton, J.; Douglas, T.; Young, M. Isolation of Viruses from High Temperature Environments. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 501, pp. 43–54. ISBN 978-1-58829-682-5. [Google Scholar]
- FastX-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 17 March 2017).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. ArXiv14015130 Q-Bio 2014. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Lerat, E.; Daubin, V.; Moran, N.A. From Gene Trees to Organismal Phylogeny in Prokaryotes: The Case of the γ-Proteobacteria. PLoS Biol. 2003, 1, e19. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Korber, B. Computational Analysis of HIV Molecular Sequences. In HIV Signature and Sequence Variation Analysis; Rodrigo, A.G., Learn, G.H., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000; pp. 55–72. ISBN 978-0-7923-7994-2. [Google Scholar]
- HIV Sequence Database. Available online: www.hiv.lanl.gov (accessed on 17 March 2017).
- Held, N.L.; Herrera, A.; Cadillo-Quiroz, H.; Whitaker, R.J. CRISPR Associated Diversity within a Population of Sulfolobus islandicus. PLoS ONE 2010, 5, e12988. [Google Scholar] [CrossRef] [PubMed]
- Cadillo-Quiroz, H.; Didelot, X.; Held, N.L.; Herrera, A.; Darling, A.; Reno, M.L.; Krause, D.J.; Whitaker, R.J. Patterns of Gene Flow Define Species of Thermophilic Archaea. PLoS Biol. 2012, 10, e1001265. [Google Scholar] [CrossRef] [PubMed]
- Reno, M.L.; Held, N.L.; Fields, C.J.; Burke, P.V.; Whitaker, R.J. Biogeography of the Sulfolobus islandicus pan-genome. Proc. Natl. Acad. Sci. USA 2009, 106, 8605–8610. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed]
- Manica, A.; Zebec, Z.; Steinkellner, J.; Schleper, C. Unexpectedly broad target recognition of the CRISPR-mediated virus defence system in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2013, 41, 10509–10517. [Google Scholar] [CrossRef] [PubMed]
- Szymczyna, B.R.; Taurog, R.E.; Young, M.J.; Snyder, J.C.; Johnson, J.E.; Williamson, J.R. Synergy of NMR, Computation, and X-ray Crystallography for Structural Biology. Struct. (Lond. Engl. 1993) 2009, 17, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, G.; Shah, S.A.; Bize, A.; Reitberger, W.; Reuter, M.; Phan, H.; Briegel, A.; Rachel, R.; Garrett, R.A.; Prangishvili, D. Stygiolobus Rod-Shaped Virus and the Interplay of Crenarchaeal Rudiviruses with the CRISPR Antiviral System. J. Bacteriol. 2008, 190, 6837–6845. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; White, M.F.; Forterre, P.; Prangishvili, D. Postcards from the Edge. In Advances in Virus Research; Elsevier: San Diego, CA, USA, 2012; Volume 82, pp. 33–62. ISBN 978-0-12-394621-8. [Google Scholar]
- Snyder, J.C.; Brumfield, S.K.; Peng, N.; She, Q.; Young, M.J. Sulfolobus Turreted Icosahedral Virus c92 Protein Responsible for the Formation of Pyramid-Like Cellular Lysis Structures. J. Virol. 2011, 85, 6287–6292. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Prangishvili, D.; Hendrix, R.W.; Bamford, D.H. Genomics of Bacterial and Archaeal Viruses: Dynamics within the Prokaryotic Virosphere. Microbiol. Mol. Biol. Rev. 2011, 75, 610–635. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Wang, C.; Upton, C. Poxviruses: Past, present and future. Virus Res. 2006, 117, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Samson, J.E.; Magadán, A.H.; Sabri, M.; Moineau, S. Revenge of the phages: Defeating bacterial defences. Nat. Rev. Microbiol. 2013, 11, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Denomy, J.; Pawluk, A.; Maxwell, K.L.; Davidson, A.R. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 2013, 493, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Denomy, J.; Garcia, B.; Strum, S.; Du, M.; Rollins, M.F.; Hidalgo-Reyes, Y.; Wiedenheft, B.; Maxwell, K.L.; Davidson, A.R. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 2015, 526, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Held, N.L.; Herrera, A.; Whitaker, R.J. Reassortment of CRISPR repeat-spacer loci in Sulfolobus islandicus. Environ. Microbiol. 2013, 15, 3065–3076. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bautista, M.A.; Black, J.A.; Youngblut, N.D.; Whitaker, R.J. Differentiation and Structure in Sulfolobus islandicus Rod-Shaped Virus Populations. Viruses 2017, 9, 120. https://doi.org/10.3390/v9050120
Bautista MA, Black JA, Youngblut ND, Whitaker RJ. Differentiation and Structure in Sulfolobus islandicus Rod-Shaped Virus Populations. Viruses. 2017; 9(5):120. https://doi.org/10.3390/v9050120
Chicago/Turabian StyleBautista, Maria A., Jesse A. Black, Nicholas D. Youngblut, and Rachel J. Whitaker. 2017. "Differentiation and Structure in Sulfolobus islandicus Rod-Shaped Virus Populations" Viruses 9, no. 5: 120. https://doi.org/10.3390/v9050120
APA StyleBautista, M. A., Black, J. A., Youngblut, N. D., & Whitaker, R. J. (2017). Differentiation and Structure in Sulfolobus islandicus Rod-Shaped Virus Populations. Viruses, 9(5), 120. https://doi.org/10.3390/v9050120