
Rotavirus Genomic RNA Complex Forms via Specific RNA–RNA Interactions: Disruption of RNA Complex Inhibits Virus Infectivity


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Sequence-Independent Cloning of RRV Segments
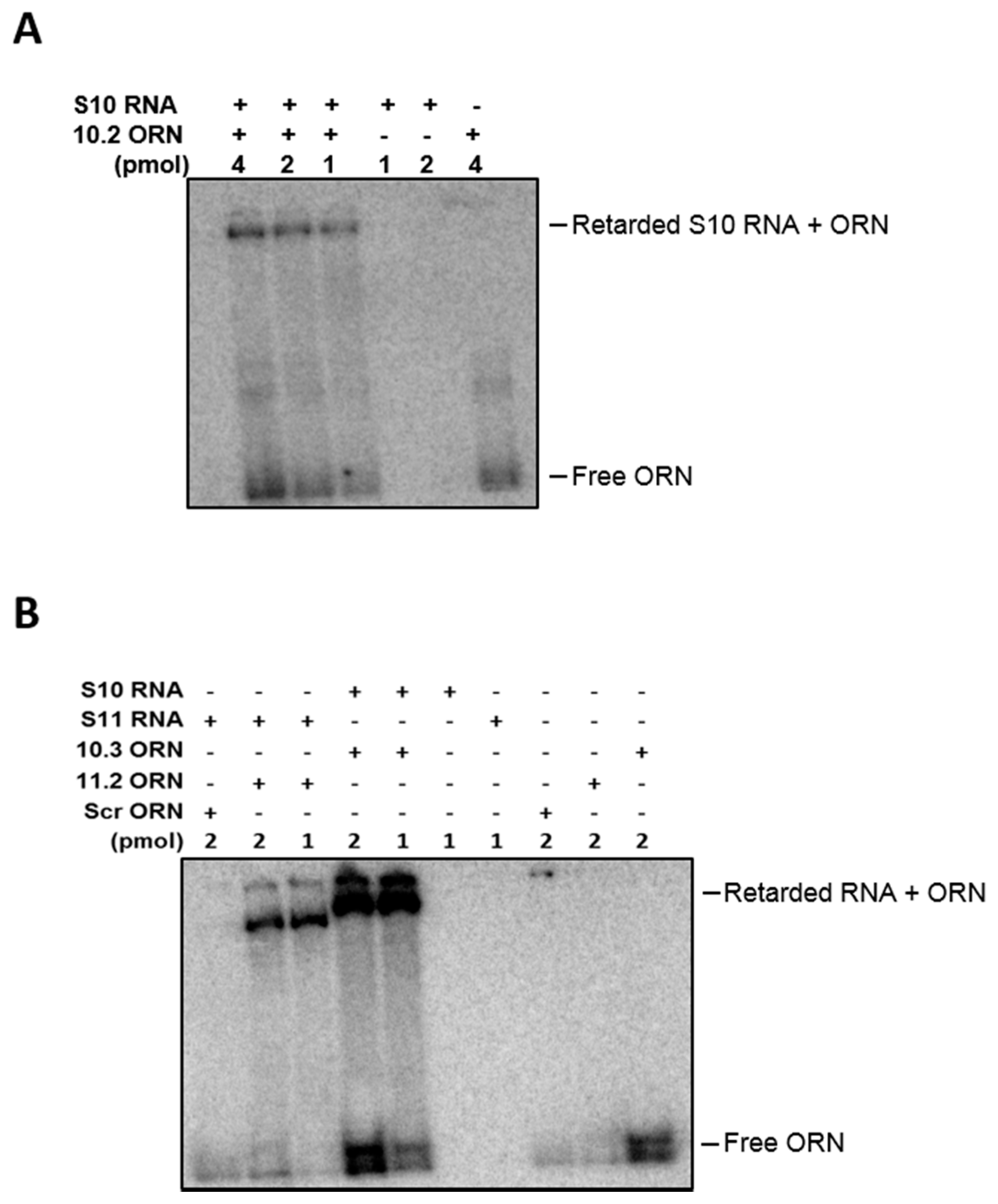
2.3. Electrophoretic Mobility Shift Assay (EMSA) for RNA–RNA Interaction
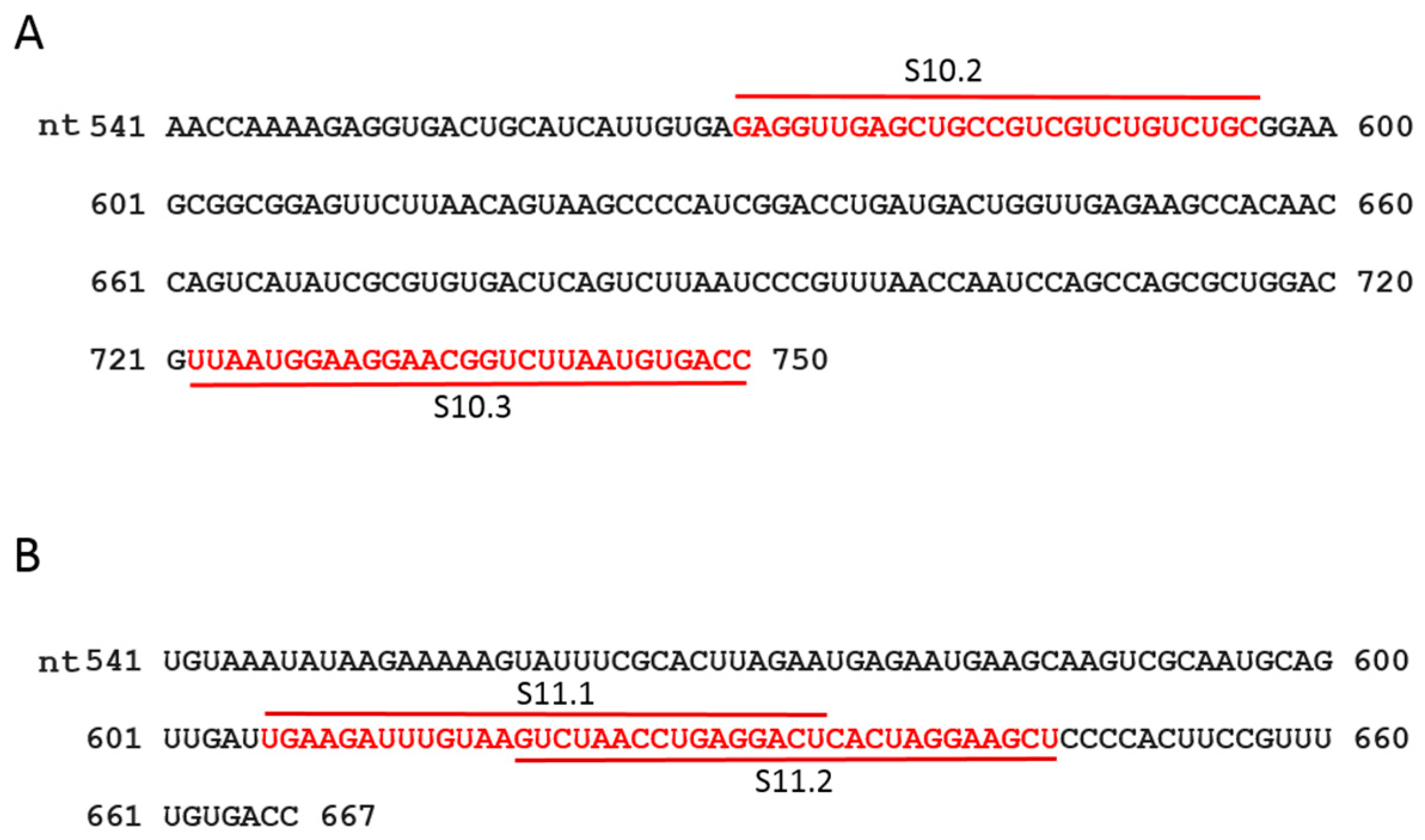
2.4. Design of Antisense Oligoribonucleotides Based on Predicted ssRNA Structures
2.5. Oligoribonucleotide Interference Assay
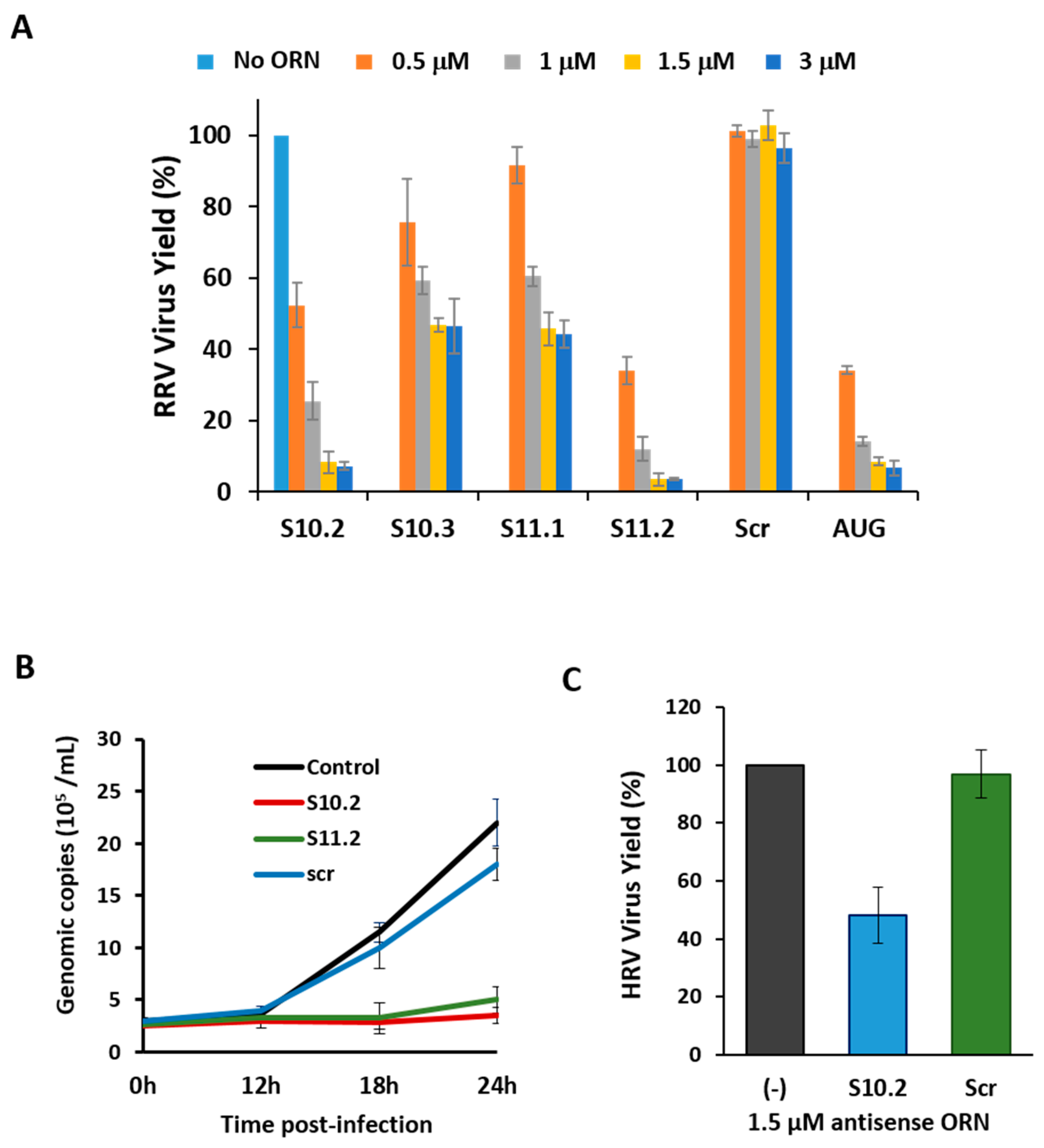
2.6. Virus Replication Inhibition Assay
2.7. Mutagenesis of RV Segment S10
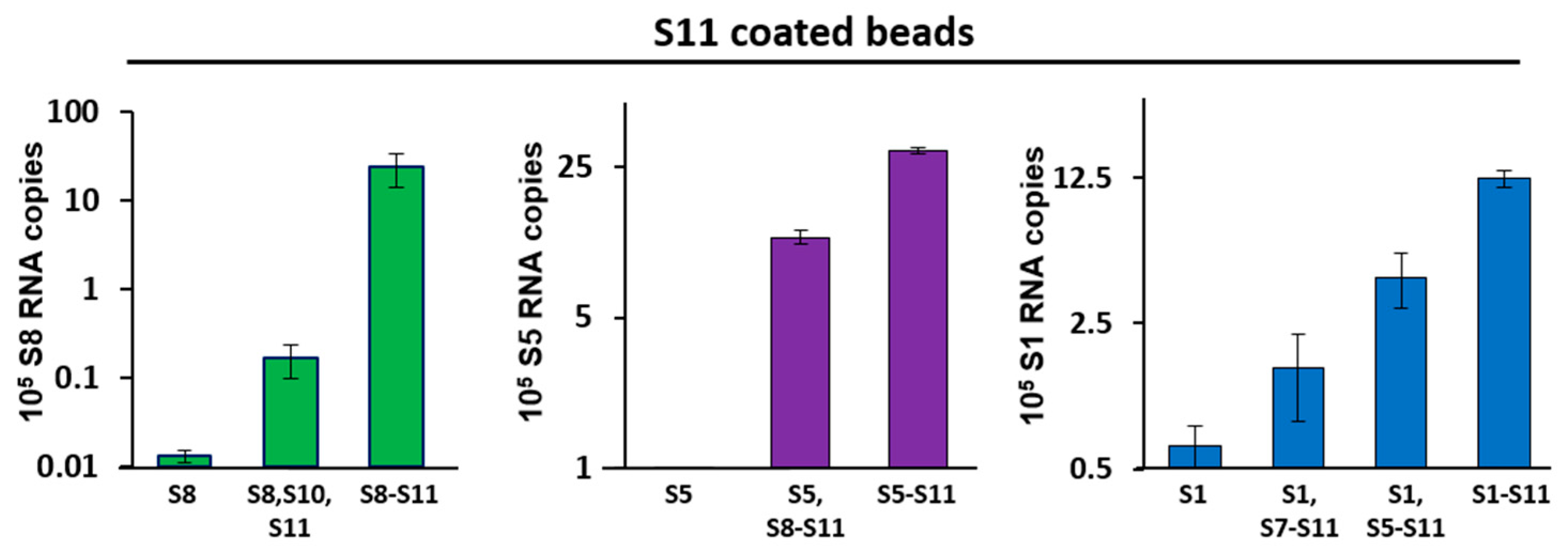
2.8. Agarose Beads Pull-Down RNA–RNA Interaction Assay
2.9. qRT-PCR
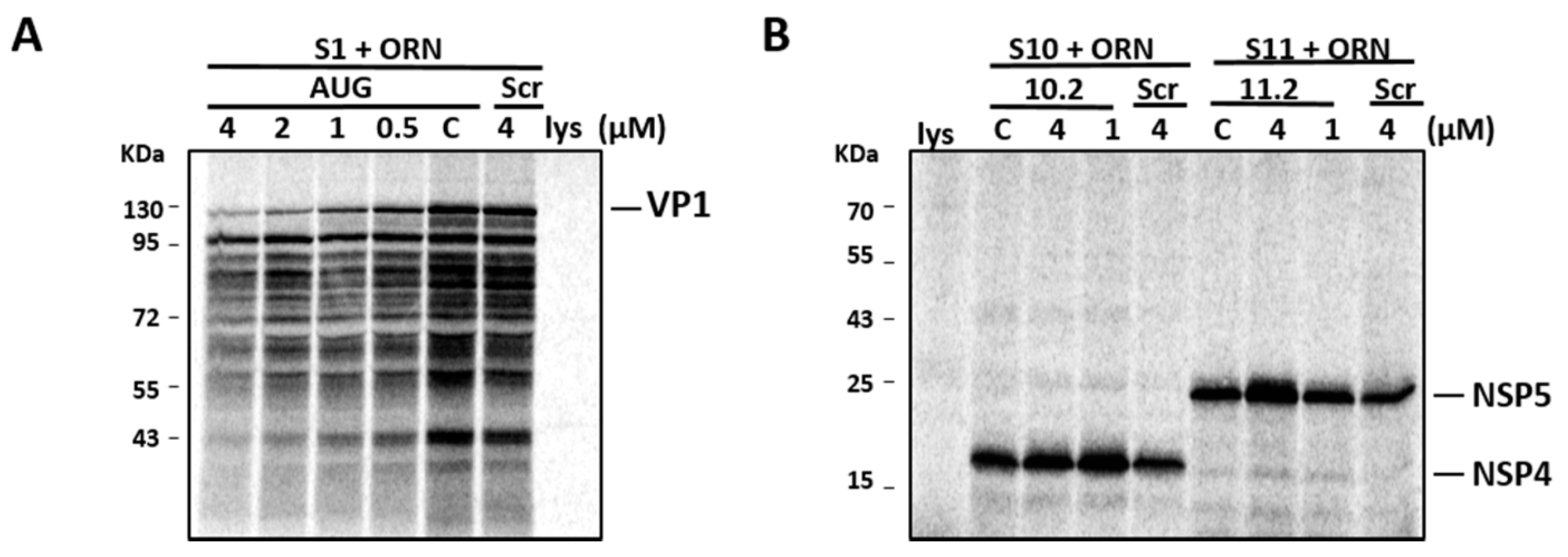
2.10. In Vitro Translation of RV Transcripts in the Presence of Oligoribonucleotides
3. Results
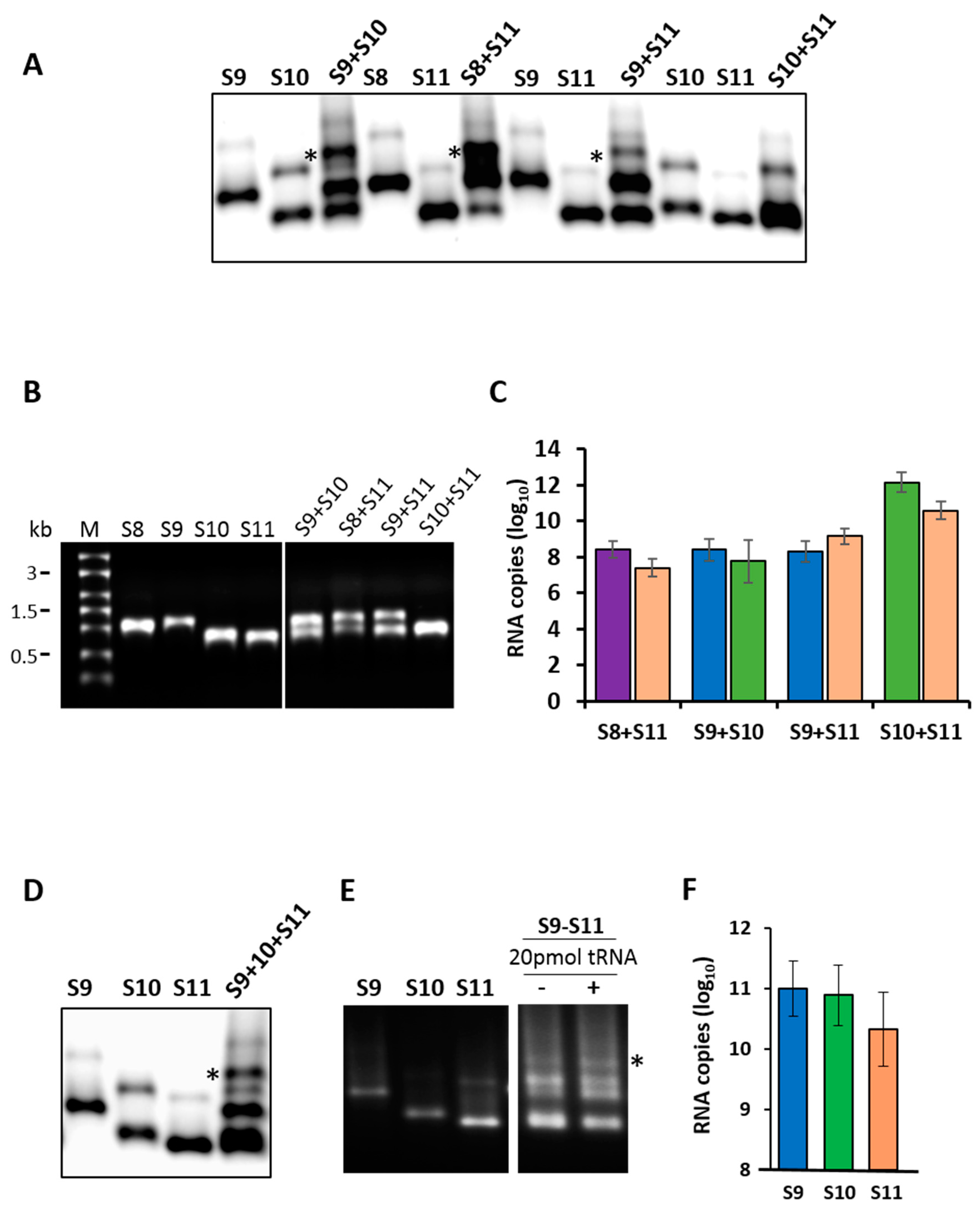
3.1. Determination of Intermolecular RNA–RNA Interactions between RRV Segments
3.2. Pull-Down Assay Suggests Networking between RNA Segments of Different Sizes
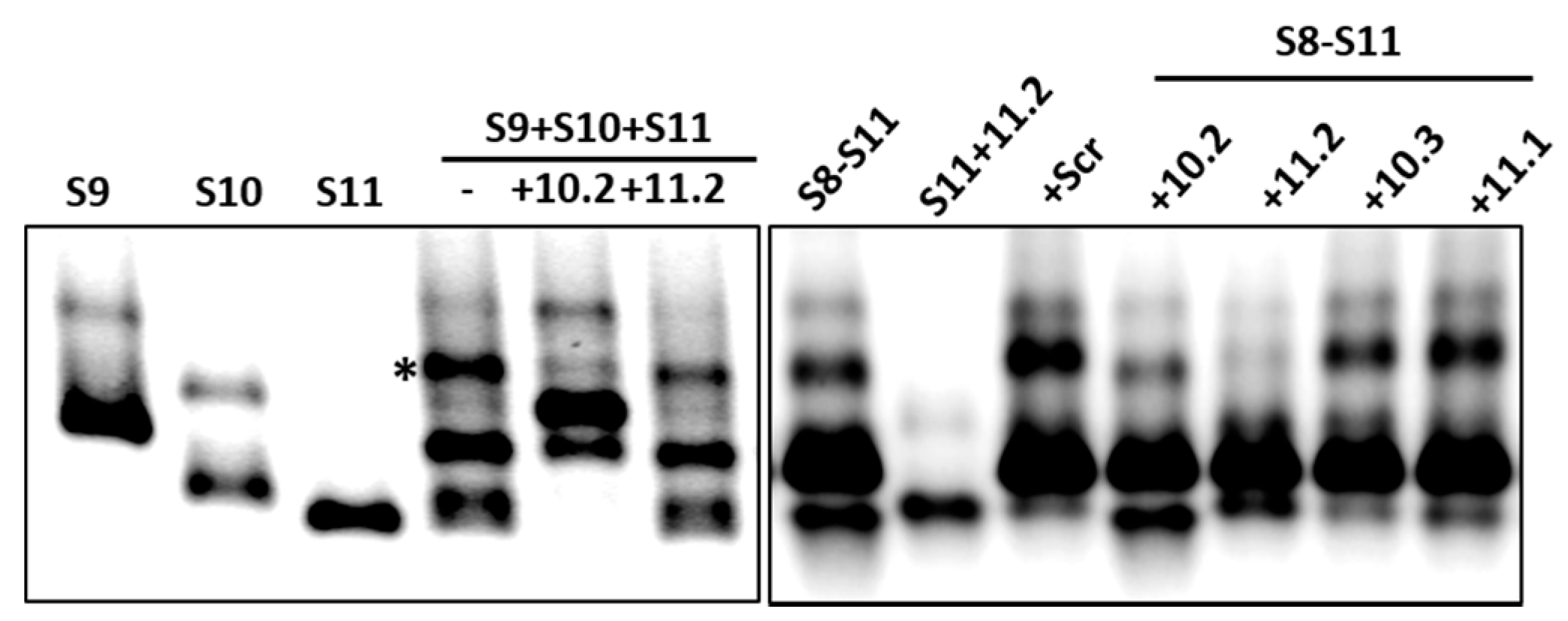
3.3. ssRNA Segment Interactions and Their Interruptions by Specific Oligoribonucleotides
3.4. Inhibition of Virus Replication with Oligoribonucleotides 3′-UTRs
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pesavento, J.B.; Crawford, S.E.; Estes, M.K.; Prasad, B.V. Rotavirus proteins: Structure and assembly. Curr. Top. Microbiol. Immunol. 2006, 309, 189–219. [Google Scholar] [PubMed]
- Estes, M.K.; Cohen, J. Rotavirus gene structure and function. Microbiol. Rev. 1989, 53, 410–449. [Google Scholar] [PubMed]
- Patton, J.T. Structure and function of the rotavirus RNA-binding proteins. J. Gen. Virol. 1995, 76, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.; Prasad, B.V. Rotavirus cell entry. Curr. Top. Microbiol. Immunol. 2010, 343, 121–148. [Google Scholar] [PubMed]
- Arias, C.F.; Isa, P.; Guerrero, C.A.; Mendez, E.; Zarate, S.; Lopez, T.; Espinosa, R.; Romero, P.; Lopez, S. Molecular biology of rotavirus cell entry. Arch. Med. Res. 2002, 33, 356–361. [Google Scholar] [CrossRef]
- Patton, J.T.; Silvestri, L.S.; Tortorici, M.A.; Vasquez-Del Carpio, R.; Taraporewala, Z.F. Rotavirus genome replication and morphogenesis: Role of the viroplasm. Curr. Top. Microbiol. Immunol. 2006, 309, 169–187. [Google Scholar] [PubMed]
- Cohen, J.; Charpilienne, A.; Chilmonczyk, S.; Estes, M.K. Nucleotide sequence of bovine rotavirus gene 1 and expression of the gene product in baculovirus. Virology 1989, 171, 131–140. [Google Scholar] [CrossRef]
- Zeng, C.Q.; Wentz, M.J.; Cohen, J.; Estes, M.K.; Ramig, R.F. Characterization and replicase activity of double-layered and single-layered rotavirus-like particles expressed from baculovirus recombinants. J. Virol. 1996, 70, 2736–2742. [Google Scholar] [PubMed]
- Lawton, J.A.; Estes, M.K.; Prasad, B.V.V. Mechanism of genome transcription in segmented dsRNA viruses. Adv. Virus Res. 2000, 55, 185–229. [Google Scholar] [PubMed]
- Trask, S.D.; McDonald, S.M.; Patton, J.T. Structural insights into the coupling of virion assembly and rotavirus replication. Nat. Rev. Microbiol. 2012, 10, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Manktelow, E.; von Kirchbach, J.C.; Gog, J.R.; Desselberger, U.; Lever, A.M. Genomic analysis of codon, sequence and structural conservation with selective biochemical-structure mapping reveals highly conserved and dynamic structures in rotavirus RNAs with potential cis-acting functions. Nucleic Acids Res. 2010, 38, 7718–7735. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hong, Y.; Parslow, T.G. Cis-acting packaging signals in the influenza virus PB1, PB2, and PA genomic RNA segments. J. Virol. 2005, 79, 10348–10355. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, C.; Sung, P.Y.; Celma, C.C.; Roy, P. Structural constraints in the packaging of bluetongue virus genomic segments. J. Gen. Virol. 2014, 95, 2240–2250. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; McCrae, M.A.; Boyce, P.; Kim, J.T. Inter-segment complementarity in orbiviruses: A driver for co-ordinated genome packaging in the reoviridae? J. Gen. Virol. 2016, 97, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Roner, M.R.; Steele, B.G. Localizing the reovirus packaging signals using an engineered m1 and s2 ssRNA. Virology 2007, 358, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Gavazzi, C.; Yver, M.; Isel, C.; Smyth, R.P.; Rosa-Calatrava, M.; Lina, B.; Moules, V.; Marquet, R. A functional sequence-specific interaction between influenza a virus genomic rna segments. Proc. Natl. Acad. Sci. USA 2013, 110, 16604–16609. [Google Scholar] [CrossRef] [PubMed]
- Fournier, E.; Moules, V.; Essere, B.; Paillart, J.C.; Sirbat, J.D.; Isel, C.; Cavalier, A.; Rolland, J.P.; Thomas, D.; Lina, B.; et al. A supramolecular assembly formed by influenza a virus genomic RNA segments. Nucleic Acids Res. 2012, 40, 2197–2209. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, T., Jr.; Sung, P.Y.; Roy, P. Disruption of specific RNA-RNA interactions in a double-stranded RNA virus inhibits genome packaging and virus infectivity. PLoS Pathog. 2015, 11, e1005321. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.Y.; Roy, P. Sequential packaging of rna genomic segments during the assembly of bluetongue virus. Nucleic Acids Res. 2014, 42, 13824–13838. [Google Scholar] [CrossRef] [PubMed]
- Poranen, M.M.; Bamford, D.H. Packaging and replication regulation revealed by chimeric genome segments of double-stranded RNA bacteriophage Phi6. RNA 1999, 5, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Noda, T. Genome packaging mechanism of influenza a virus. Yakugaku Zasshi 2015, 135, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.M.; Patton, J.T. Assortment and packaging of the segmented rotavirus genome. Trends Microbiol. 2011, 19, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, T., Jr.; AlShaikhahmed, K.; Roy, P. Generation of infectious rna complexes in orbiviruses: RNA-RNA interactions of genomic segments. Oncotarget 2016, 7, 72559–72570. [Google Scholar] [CrossRef] [PubMed]
- Periz, J.; Celma, C.; Jing, B.; Pinkney, J.N.; Roy, P.; Kapanidis, A.N. Rotavirus mRNAs are released by transcript-specific channels in the double-layered viral capsid. Proc. Natl. Acad. Sci. USA 2013, 110, 12042–12047. [Google Scholar] [CrossRef] [PubMed]
- Maan, S.; Rao, S.; Maan, N.S.; Anthony, S.J.; Attoui, H.; Samuel, A.R.; Mertens, P.P. Rapid cDNA synthesis and sequencing techniques for the genetic study of bluetongue and other dsRNA viruses. J. Virol. Methods 2007, 143, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.; Martinez-Salas, E. Long-range RNA interactions between structural domains of the aphthovirus internal ribosome entry site (IRES). RNA 1999, 5, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Helene, C.; Toulme, J.J. Specific regulation of gene expression by antisense, sense and antigene nucleic acids. Biochim. Biophys. Acta 1990, 1049, 99–125. [Google Scholar] [CrossRef]
- Broering, R.; Real, C.I.; John, M.J.; Jahn-Hofmann, K.; Ickenstein, L.M.; Kleinehr, K.; Paul, A.; Gibbert, K.; Dittmer, U.; Gerken, G.; et al. Chemical modifications on sirnas avoid toll-like-receptor-mediated activation of the hepatic immune system in vivo and in vitro. Int. Immunol. 2014, 26, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.H.; Bochkareva, E.; Bochkarev, A.; Mou, T.C.; Gray, D.M. 2′-O-methyl-modified phosphorothioate antisense oligonucleotides have reduced non-specific effects in vitro. Nucleic Acids Res. 2004, 32, 2008–2016. [Google Scholar] [CrossRef] [PubMed]
- NCBI-BLAST. Available online: http://blast.ncbi.nlm.nih.gov/ (accessed on 26 June 2017).
- Mfold. Available online: http://rna.tbi.univie.ac.at/ (accessed on 26 June 2017).
- RNAfold. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi (accessed on 26 June 2017).
- OligoAnalyzer. Available online: http://eu.idtdna.com/calc/analyzer (accessed on 26 June 2017).
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar] [PubMed]
- Weiner, M.P.; Costa, G.L.; Schoettlin, W.; Cline, J.; Mathur, E.; Bauer, J.C. Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene 1994, 151, 119–123. [Google Scholar] [CrossRef]
- Boyce, M.; Celma, C.C.; Roy, P. Development of reverse genetics systems for bluetongue virus: Recovery of infectious virus from synthetic RNA transcripts. J. Virol. 2008, 82, 8339–8348. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; McCrae, M.A. Rapid mapping of functional cis-acting rna elements by recovery of virus from a degenerate RNA population: Application to genome segment 10 of bluetongue virus. J. Gen. Virol. 2015, 96, 3072–3082. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.K.; Greenberg, H.B. Rotaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- McDonald, S.M.; Nelson, M.I.; Turner, P.E.; Patton, J.T. Reassortment in segmented RNA viruses: Mechanisms and outcomes. Nat. Rev. Microbiol. 2016, 14, 448–460. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORN | Sequence (2′-O-Methyl Modified) | Position |
|---|---|---|
| S1 AUG | 5′-GAUUAGAUUAUACUUCCCCAUUGUAUAGC-3′ (29 nts) | 28–56 |
| S10.2 | 5′-GCAGACAGACGACGGCAGCUCAACCUC-3′ (27 nts) | 570–596 |
| S10.3 | 5′-GGUCACAUUAAGACCGUUCCUUCCAUUAA-3′ (29 nts) | 722–750 |
| S11.1 | 5′-AGUCCUCAGGUUAGACUUACAAAUCUUCA-3′ (29 nts) | 606–625 |
| S11.2 | 5′-AGCUUCCUAGUGAGUCCUCAGGUUAGAC-3′ (28 nts) | 619–646 |
| Scr | 5′-UCGUGCAAUACUAGGAUCACUAUUACAU-3′ (28 nts) | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fajardo Jr., T.; Sung, P.-Y.; Celma, C.C.; Roy, P. Rotavirus Genomic RNA Complex Forms via Specific RNA–RNA Interactions: Disruption of RNA Complex Inhibits Virus Infectivity. Viruses 2017, 9, 167. https://doi.org/10.3390/v9070167
Fajardo Jr. T, Sung P-Y, Celma CC, Roy P. Rotavirus Genomic RNA Complex Forms via Specific RNA–RNA Interactions: Disruption of RNA Complex Inhibits Virus Infectivity. Viruses. 2017; 9(7):167. https://doi.org/10.3390/v9070167
Chicago/Turabian StyleFajardo Jr., Teodoro, Po-Yu Sung, Cristina C. Celma, and Polly Roy. 2017. "Rotavirus Genomic RNA Complex Forms via Specific RNA–RNA Interactions: Disruption of RNA Complex Inhibits Virus Infectivity" Viruses 9, no. 7: 167. https://doi.org/10.3390/v9070167
APA StyleFajardo Jr., T., Sung, P. -Y., Celma, C. C., & Roy, P. (2017). Rotavirus Genomic RNA Complex Forms via Specific RNA–RNA Interactions: Disruption of RNA Complex Inhibits Virus Infectivity. Viruses, 9(7), 167. https://doi.org/10.3390/v9070167