
Crystal Structure of the Carboxy-Terminal Region of the Bacteriophage T4 Proximal Long Tail Fiber Protein Gp34

 and
and
Abstract
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Structure Solution
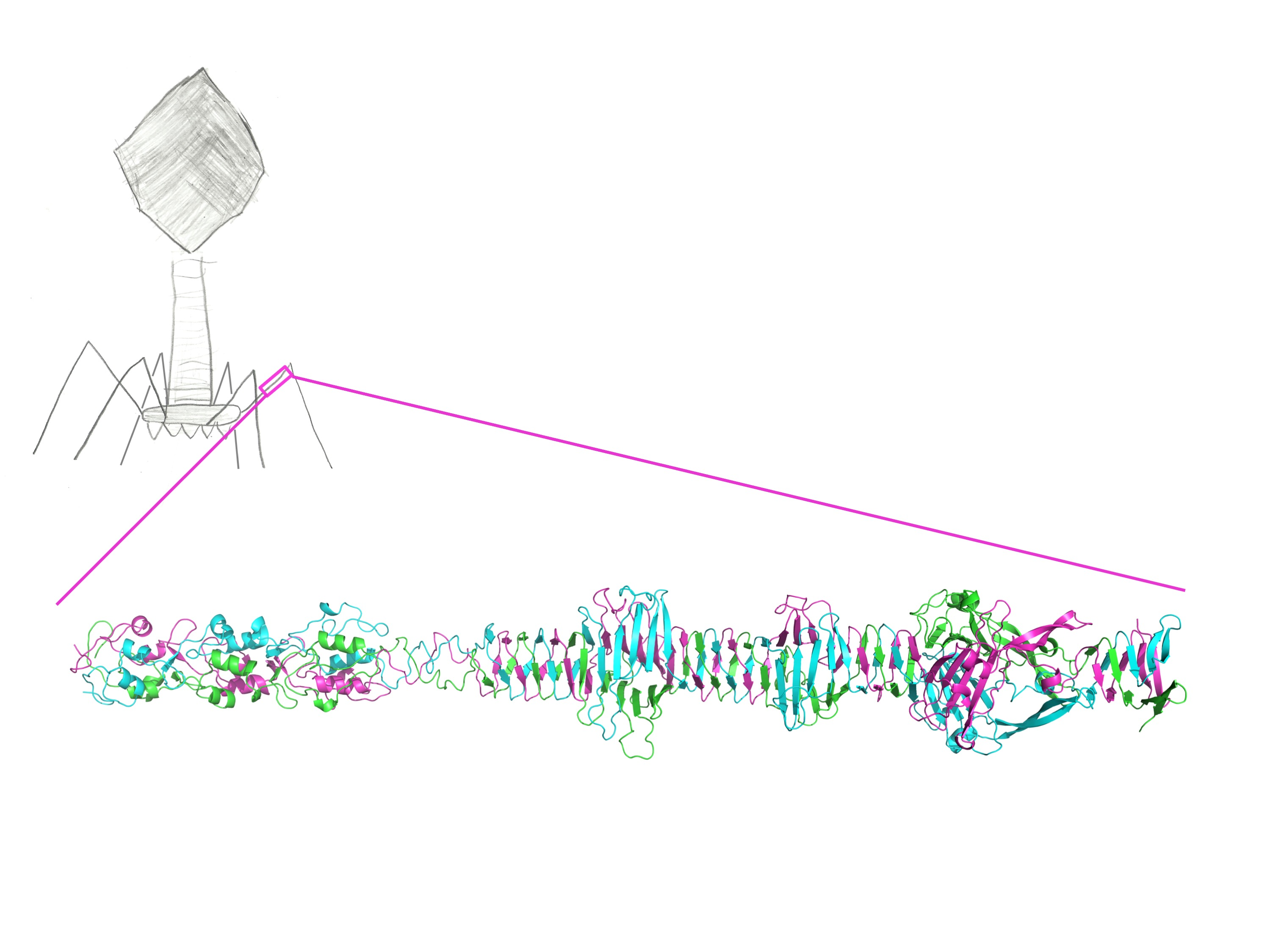
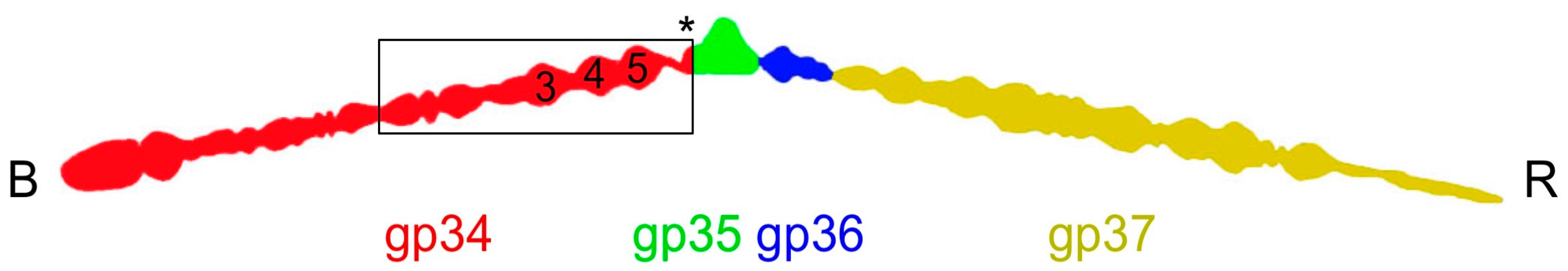
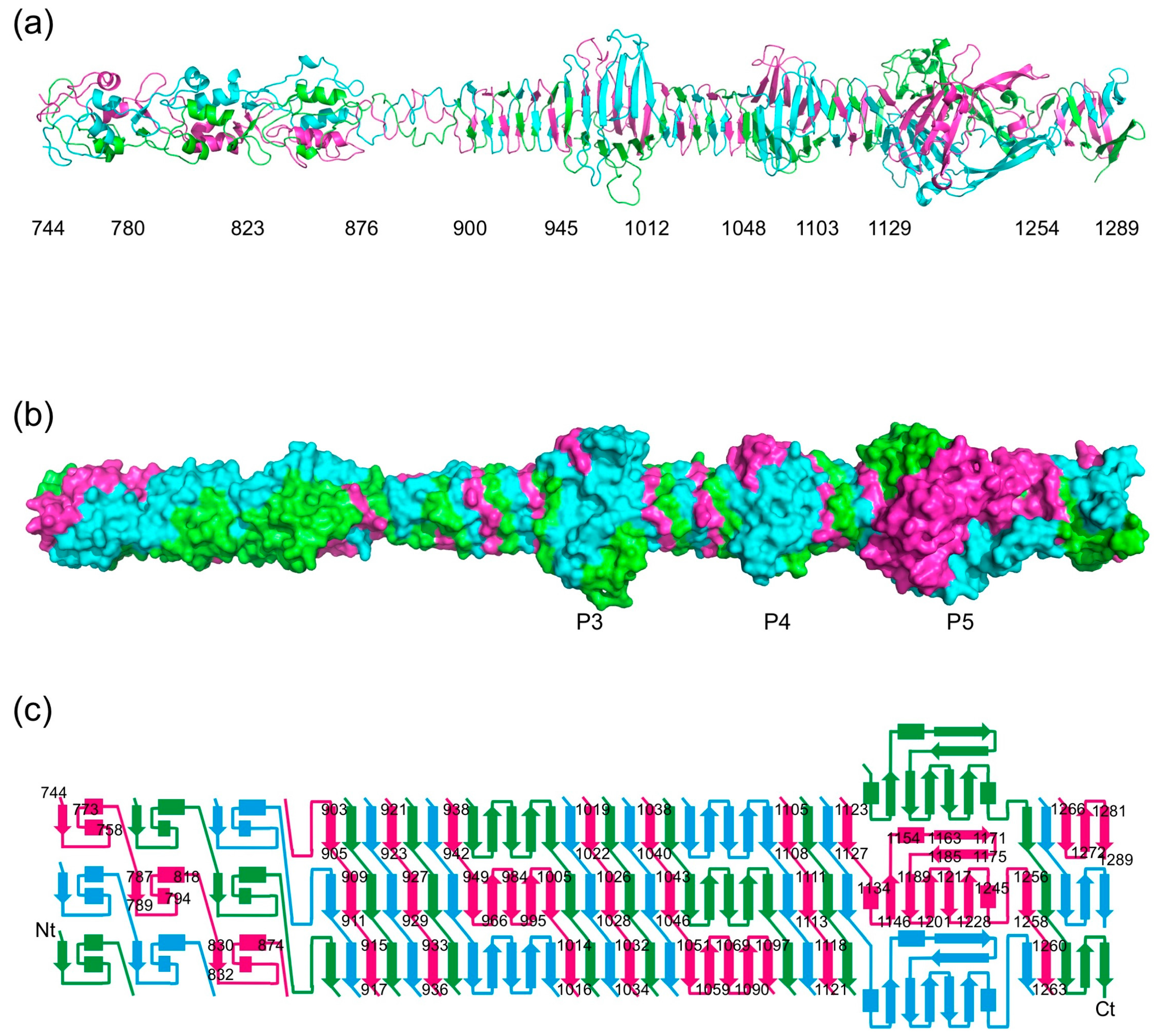
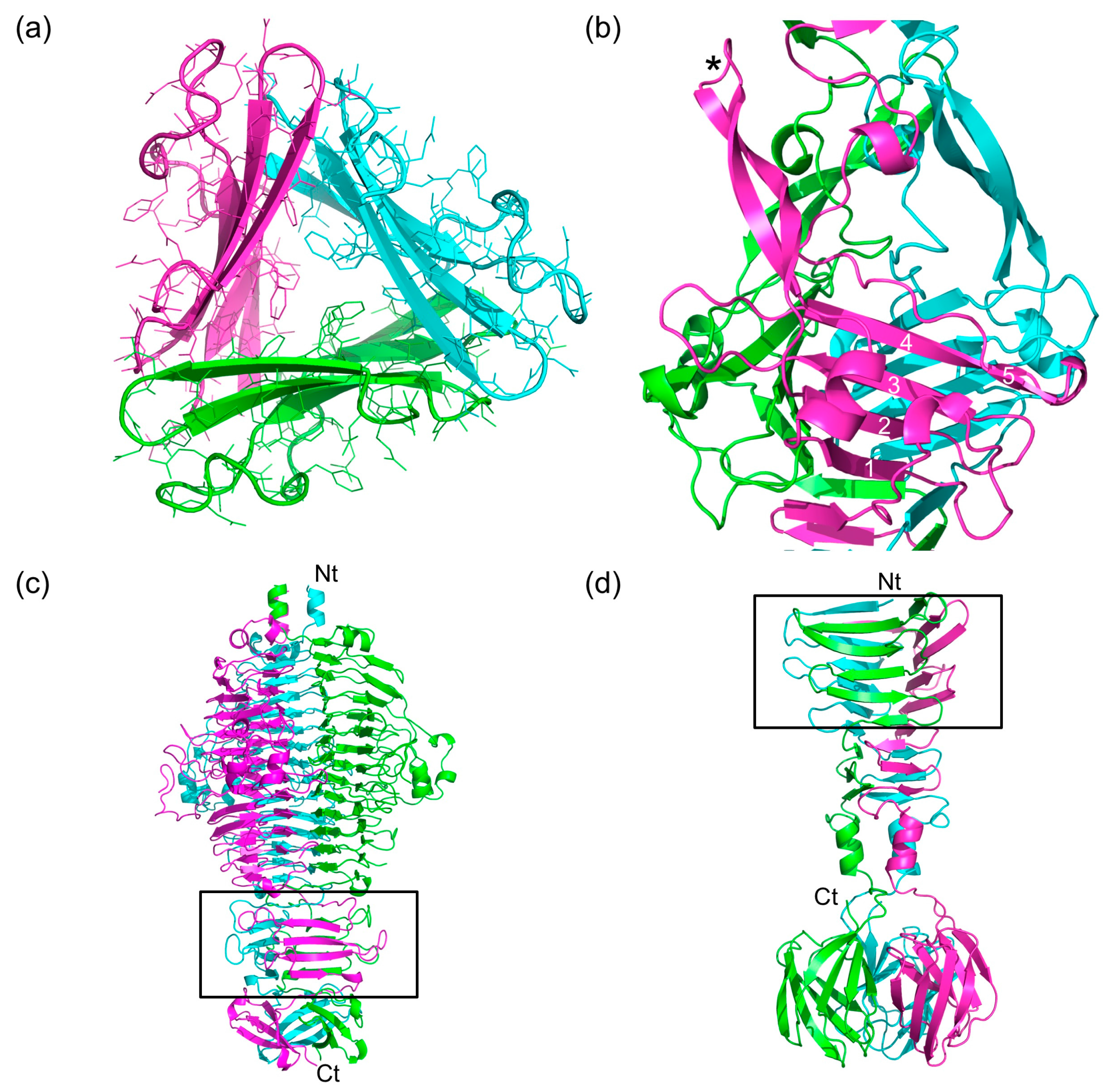
3.2. Overall Structure
3.3. Stability of the Trimer
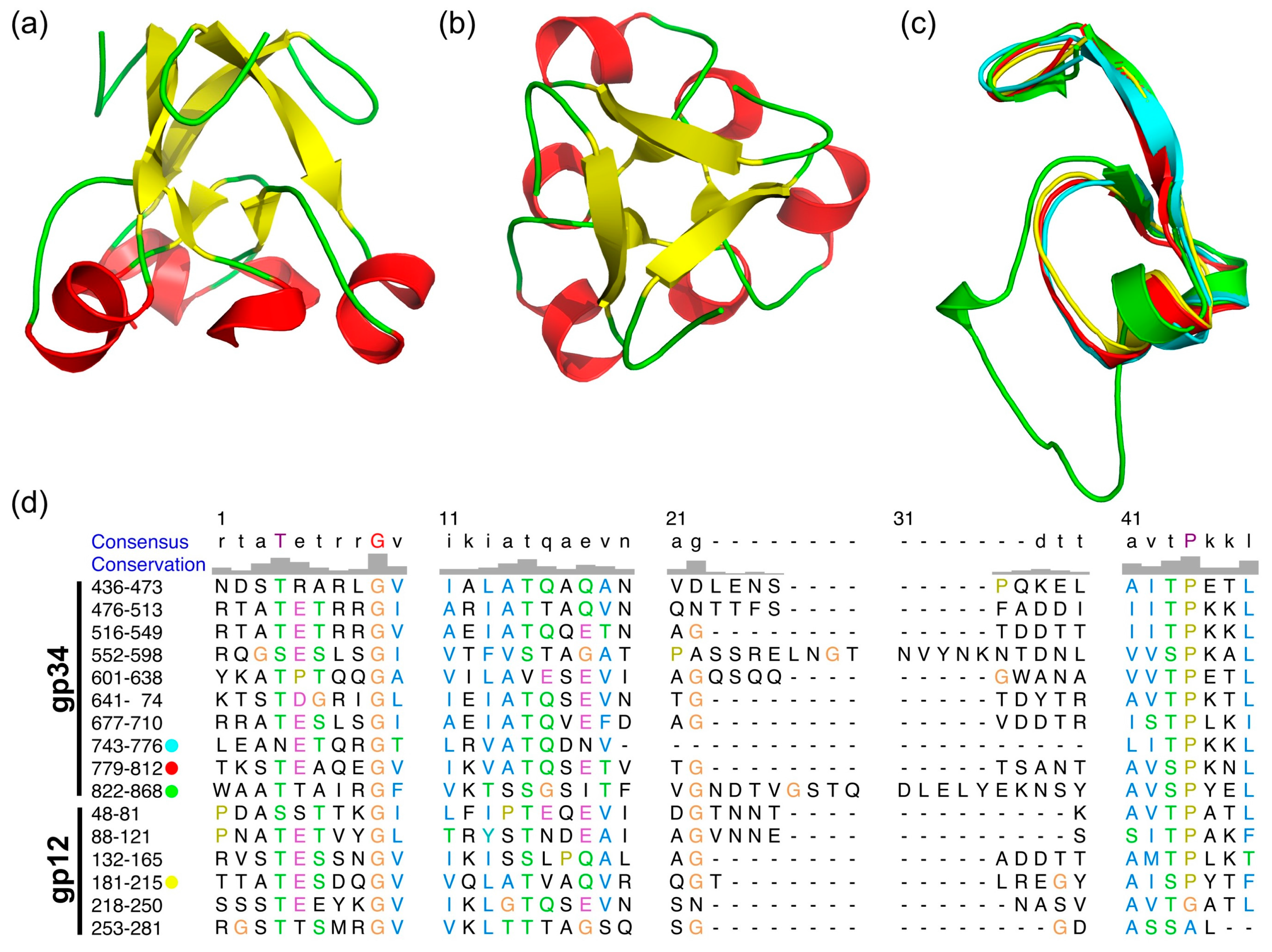
3.4. The α-Helix-Containing Domains
3.5. The Triple β-Helix
3.6. The P3, P4, and P5 Domains
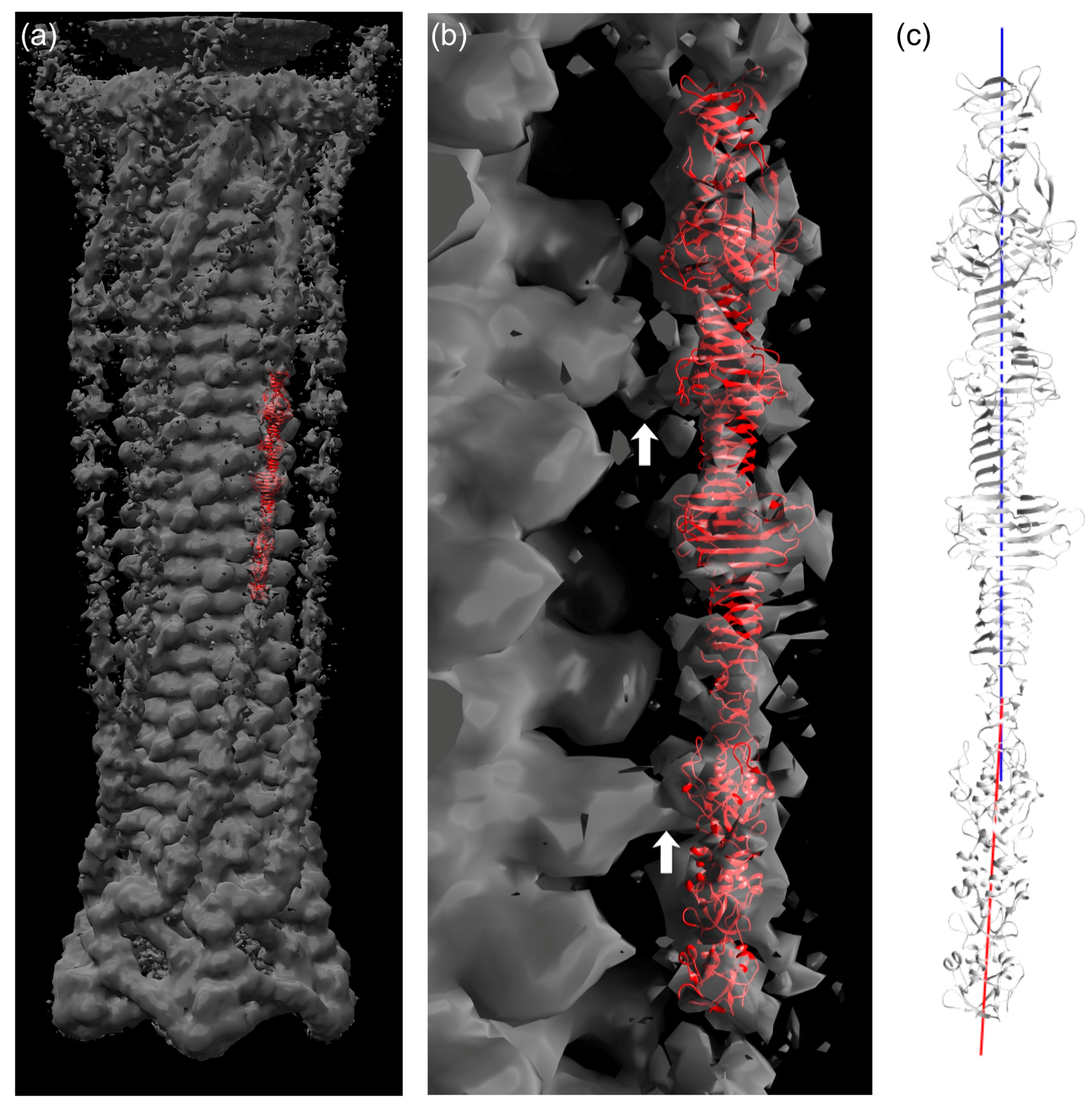
3.7. Fitting of the Crystal Structure in an EM Map of T4 Phage
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cairns, J.; Stent, G.S.; Watson, J.D. Phage and the Origins of Molecular Biology, the Centennial Edition; Cold Spring Harbor laboratory Press: New York, NY, USA, 2007; ISBN 9780879698003. [Google Scholar]
- Canchaya, C.; Fournous, G.; Chibani-Chennoufi, S.; Dillmann, M.L.; Brüssow, H. Phage as agents of lateral gene transfer. Curr. Opin. Microbiol. 2003, 6, 417–424. [Google Scholar] [CrossRef]
- Brüssow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.R.; March, J.B. Bacteriophages and biotechnology: Vaccines, gene therapy and antibacterials. Trends Biotechnol. 2006, 24, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. Bacteriophage observations and evolution. Res. Microbiol. 2003, 154, 245–251. [Google Scholar] [CrossRef]
- Ackermann, H.W.; Prangishvili, D. Prokaryote viruses studied by electron microscopy. Arch. Virol. 2012, 157, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Karam, J.D.; Drake, J.W.; Kreuzer, K.N.; Mosig, G.; Hall, D.H.; Eiserling, F.A.; Black, L.W.; Spicer, E.K.; Kutter, E.; Carlson, K.; et al. Molecular Biology of Bacteriophage T4; American Society for Microbiology: Washington, DC, USA, 1994; ISBN 9781555810641. [Google Scholar]
- Leiman, P.G.; Arisaka, F.; van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the T4 tail and tail fibers. Virol. J. 2010, 7, 355. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.I.; Prokhorov, N.S.; Guerrero-Ferreira, R.C.; Shneider, M.M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P.G. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 2016, 533, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc. Natl. Acad. Sci. USA 2015, 112, E4919–E4928. [Google Scholar] [CrossRef] [PubMed]
- Riede, I. Receptor specificity of the short tail fibres (gp12) of T-even type Escherichia coli phages. Mol. Gen. Genet. 1987, 206, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Kostyuchenko, V.A.; Leiman, P.G.; Chipman, P.R.; Kanamaru, S.; van Raaij, M.J.; Arisaka, F.; Mesyanzhinov, V.V.; Rossmann, M.G. Three-dimensional structure of bacteriophage T4 baseplate. Nat. Struct. Biol. 2003, 10, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, M.E.; Wall, J.S.; Simon, M.N.; Conway, J.F.; Steven, A.C. Stoichiometry and domainal organization of the long tail-fiber of bacteriophage T4 A hinged viral adhesin. J. Mol. Biol. 1996, 260, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Granell, M.; Namura, M.; Alvira, S.; Garcia-Doval, C.; Singh, A.K.; Gutsche, I.; van Raaij, M.J.; Kanamaru, S. Crystallization of the carboxy-terminal region of the bacteriophage T4 proximal long tail fibre protein gp34. Acta Cryst. Sect. F 2014, 70, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Bartual, S.G.; Otero, J.M.; Garcia-Doval, C.; Llamas-Saiz, A.L.; Kahn, R.; Fox, G.C.; van Raaij, M.J. Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc. Natl. Acad. Sci. USA 2010, 107, 20287–20292. [Google Scholar] [CrossRef] [PubMed]
- Hashemolhosseini, S.; Stierhof, Y.D.; Hindennach, I.; Henning, U. Characterization of the helper proteins for the assembly of tail fibers of coliphages T4 and λ. J. Bacteriol. 1996, 178, 6258–6265. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Iwabuchi, N.; Matsui, T.; Hirota, K.; Kidokoro, S.; Arai, M.; Kuwajima, K.; Schuck, P.; Arisaka, F. Reversible and fast association equilibria of a molecular chaperone, gp57A, of bacteriophage T4. Biophys. J. 2003, 85, 2606–2618. [Google Scholar] [CrossRef]
- Bartual, S.G.; Garcia-Doval, C.; Alonso, J.; Schoehn, G.; van Raaij, M.J. Two-chaperone assisted soluble expression and purification of the bacteriophage T4 long tail fibre protein gp37. Protein Expr. Purif. 2010, 70, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Van Raaij, M.J.; Schoehn, G.; Burda, M.R.; Miller, S. Crystal structure of a heat and protease-stable part of the bacteriophage T4 short tail fibre. J. Mol. Biol. 2001, 314, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Thomassen, E.; Gielen, G.; Schütz, M.; Schoehn, G.; Abrahams, J.P.; Miller, S.; van Raaij, M.J. The structure of the receptor-binding domain of the bacteriophage T4 short tail fibre reveals a knitted trimeric metal-binding fold. J. Mol. Biol. 2003, 331, 361–373. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Vonrhein, C.; Blanc, E.; Roversi, R.; Bricogne, G. Automated structure solution with Autosharp. Methods Mol. Biol. 2007, 364, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, J.P.; Leslie, A.G.W. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Cryst. Sect. D 1996, 52, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. MOLREP An automated program for molecular replacement. J. Appl. Cryst. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. Sect. D 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Headd, J.J.; Echols, N.; Afonine, P.V.; Grosse-Kunstleve, R.W.; Chen, V.B.; Moriarty, N.W.; Richardson, D.C.; Richardson, J.S.; Adams, P.D. Use of knowledge-based restraints in phenix.refine to improve macromolecular refinement at low resolution. Acta Cryst. Sect. D 2012, 68, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst. Sect. D 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Westbrook, J.D.; Feng, Z.; Sala, R.; Peisach, E.; Oldfield, T.J.; Sen, S.; Gutmanas, A.; Armstrong, D.R.; Berrisford, J.M.; et al. OneDep Unified wwPDB system for deposition, biocuration, and validation of macromolecular structures in the Protein Data Bank (PDB) archive. Structure 2017, 25, 536–545. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 1.6; Schrödinger LLC: New York, NY, USA, 2013.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera, a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E. Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res. 2015, 43, W314–W319. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.H.; Paul, R.; Dukka, K.C.; Shilling, J.D.; Lee, B. SymD webserver: A platform for detecting internally symmetric protein structures. Nucleic Acids Res. 2014, 42, W296–W300. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl. Acids. Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Aurora, R.; Rose, G.D. Helix capping. Protein Sci. 1998, 7, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, S.; Leiman, P.G.; Kostyuchenko, V.A.; Chipman, P.R.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure of the cell-puncturing device of bacteriophage T4. Nature 2002, 415, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Jabs, A.; Weiss, M.S.; Hilgenfeld, R. Non-proline cis peptide bonds in proteins. J. Mol. Biol. 1999, 286, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Steinbacher, S.; Seckler, R.; Miller, S.; Steipe, B.; Huber, R.; Reinemer, P. Crystal structure of P22 tailspike protein Interdigitated subunits in a thermostable trimer. Science 1994, 265, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Fiedler, C.; Grassl, R.; Biebl, M.; Rachel, R.; Hermo-Parrado, X.L.; Llamas-Saiz, A.L.; Seckler, R.; Miller, S.; van Raaij, M.J. Structure of the receptor-binding protein of bacteriophage Det7: A podoviral tail spike in a myovirus. J. Virol. 2008, 82, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Doval, C.; van Raaij, M.J. Structure of the receptor-binding carboxy-terminal domain of bacteriophage T7 tail fibers. Proc. Natl. Acad. Sci. USA 2012, 109, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Doval, C.; Castón, J.R.; Luque, D.; Granell, M.; Otero, J.M.; Llamas-Saiz, A.L.; Renouard, M.; Boulanger, P.; van Raaij, M.J. Structure of the receptor-binding carboxy-terminal domain of the bacteriophage T5 L-shaped tail fibre with and without its intra-molecular chaperone. Viruses 2015, 7, 6424–6440. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.C.; Dickmanns, A.; Urlaub, H; Schmitt, A.; Mühlenhoff, M.; Stummeyer, K.; Schwarzer, D.; Gerardy-Schahn, R.; Ficner, R. Crystal structure of an intramolecular chaperone mediating triple-β-helix folding. Nat. Struct. Mol. Biol. 2010, 17, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Leiman, P.G.; Li, L.; Grimes, S.; Anderson, D.L.; Rossmann, M.G. Crystallographic insights into the autocatalytic assembly mechanism of a bacteriophage tail spike. Mol. Cell 2009, 34, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Kostyuchenko, V.A.; Chipman, P.R.; Leiman, P.G.; Arisaka, F.; Mesyanzhinov, V.V.; Rossmann, M.G. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat. Struct. Mol. Biol. 2005, 12, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Van Raaij, M.J.; Mitraki, A. β-structured viral fibres: Assembly, structure and implications for materials design. Curr. Opin. Solid State Mater. Sci. 2004, 8, 151–156. [Google Scholar] [CrossRef]
- Woolfson, D.N. Building fibrous biomaterials from α-helical and collagen-like coiled-coil peptides. Biopolymers 2010, 94, 118–127. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | |||||
|---|---|---|---|---|---|
| P21-SeMet (894–1289) | P21-native (894–1289) | H32-native (894–1289) | P21-SeMet (781–1289) | P21-native (726–1289) | |
| Cell axes (a, b, c, Å) | 92.8, 76.3, 117.0 | 93.0, 76.1, 116.8 | 228.5, 228.5, 1069.5 | 92.1, 75.9, 149.1 | 107.3, 76.1, 139.9 |
| Cell angles (α, β, γ, °) | 90.0, 99.3, 90.0 | 90.0, 99.1, 90.0 | 90.0, 90.0, 120.0 | 90.0, 90.2, 90.0 | 90.0, 97.6, 90.0 |
| Beamline | Diamond I04 | ESRF ID14-1 | ESRF ID14-4 | PF, BL1A | PF, BL17A |
| Resolution range (Å) | 30–2.0 (2.11–2.00) * | 30–2.0 (2.11–2.00) | 30–3.0 (3.16–3.00) | 46.1–1.9 (2.00–1.90) | 45.2–2.89 (3.04–2.89) |
| Reflections | 109,012 (15,872) | 108,802 (15,849) | 214,066 (31,052) | 161,501 (23,029) | 48930 (6784) |
| Multiplicity | 7.4 (7.5) | 3.4 (3.4) | 4.6 (4.7) | 4.9 (4.9) | 3.3 (3.3) |
| Completeness (%) | 99.9 (99.9) | 99.9 (99.9) | 99.8 (99.8) | 99.0 (97.0) | 96.6 (91.8) |
| Mean <I/s(I)> | 12.4 (5.6) | 8.1 (3.3) | 8.4 (3.5) | 10.0 (3.2) | 9.4 (3.0) |
| Rmerge (%) † | 11.1 (30.7) | 10.6 (37.7) | 12.9 (36.1) | 11.5 (42.5) | 9.8 (43.6) |
| Wilson B (Å2) | 13.7 | 14.9 | 55.6 | 15.0 | 69.5 |
| Phasing | |||||
| Heavy atom sites ‡ | 13 Se | ||||
| Correlation coeff. (all/weak) ‡ | 51.99/29.92 | ||||
| Patterson FOM ‡ | 12.58 | ||||
| Correlation coeff. (E) ‡ | 0.505 | ||||
| R-cullis ¶ (anom., acentric) | 0.843 | ||||
| Phasing power ¶ | 0.916 | ||||
| FOM ** [cos(phase error)] (acentric/centric) | 0.2996/0.0781 | ||||
| Solvent flattening | |||||
| R-factor ** (before/after) | 0.4820/0.2120 | ||||
| Overall correlation on |E|2 ** (before/after) | 0.3246/0.8833 | ||||
| Correlation on |E|2/contrast (original/inverted) | 0.4688/0.2766 | ||||
| Refinement | |||||
| Reflections | 106789 (17167) | 106463 (17174) | 213700 (41856) | 159525 (11143) | 47006 (3004) |
| Reflections Rfree | 2205 (346) | 2209 (349) | 2999 (545) | 1957 (159) | 1911 (130) |
| R-factor †† | 0.140 (0.162) | 0.146 (0.199) | 0.226 (0.281) | 0.171 (0.244) | 0.200 (0.352) |
| R-free | 0.175 (0.207) | 0.187 (0.219) | 0.250 (0.321) | 0.208 (0.275) | 0.263 (0.408) |
| Protein/glycerol/ water/other atoms | 9091/42/1619/0 | 9101/42/1664/0 | 36241/0/0/0 | 11299/102/1822/27 | 12441/6/350/0 |
| Overall B-factor (Å2) | 20.0 | 24.9 | 51.4 | 25.9 | 64.8 |
| Ramachandran stats ‡‡ (%) | 96.5/99.8 | 96.5/99.6 | 96.2/99.9 | 96.7/99.5 | 95.5/99.9 |
| rmsd ¶¶ bonds (Å)/angles (°) | 0.013/1.3 | 0.013/1.5 | 0.005/0.9 | 0.011/1.4 | 0.011/1.5 |
| PDB code | 4UXE | 4UXF | 4UXG | 5NXF | 5NXH |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granell, M.; Namura, M.; Alvira, S.; Kanamaru, S.; Van Raaij, M.J. Crystal Structure of the Carboxy-Terminal Region of the Bacteriophage T4 Proximal Long Tail Fiber Protein Gp34. Viruses 2017, 9, 168. https://doi.org/10.3390/v9070168
Granell M, Namura M, Alvira S, Kanamaru S, Van Raaij MJ. Crystal Structure of the Carboxy-Terminal Region of the Bacteriophage T4 Proximal Long Tail Fiber Protein Gp34. Viruses. 2017; 9(7):168. https://doi.org/10.3390/v9070168
Chicago/Turabian StyleGranell, Meritxell, Mikiyoshi Namura, Sara Alvira, Shuji Kanamaru, and Mark J. Van Raaij. 2017. "Crystal Structure of the Carboxy-Terminal Region of the Bacteriophage T4 Proximal Long Tail Fiber Protein Gp34" Viruses 9, no. 7: 168. https://doi.org/10.3390/v9070168
APA StyleGranell, M., Namura, M., Alvira, S., Kanamaru, S., & Van Raaij, M. J. (2017). Crystal Structure of the Carboxy-Terminal Region of the Bacteriophage T4 Proximal Long Tail Fiber Protein Gp34. Viruses, 9(7), 168. https://doi.org/10.3390/v9070168