1. Introduction
Archaeal viruses display a high morphological and genetic diversity. They represent a separate group, distinct from bacterial and eukaryotic viruses [
1]. Amongst the unique morphologies described exclusively for archaeal viruses are spindle-, egg-, spiral- and bottle-shaped virions. Viruses infecting archaea represent the most recently discovered viruses and the limited number of viruses isolated to date is expected to represent only a small fraction of a diverse unexplored world of novel viral families [
2].
The large majority of archaeal viruses have double-stranded (ds) DNA genomes, which can be either circular or linear. The sequences of most genes encoded by these genomes yield no hits in extant databases and their functions remain largely unknown [
1,
2,
3]. Studies on the infectious biology of archaeal viruses are hampered by this low number of functionally characterized viral genes. In addition, the infection cycles of archaeal viruses are mostly unexplored. However, in recent years considerable efforts have been made to unravel the molecular mechanisms underlying infection by archaeal viruses and some have emerged as models for the study of virus–host interactions. An example of such a model is the rudivirus
Sulfolobus islandicus rod-shaped virus 2 (SIRV2). Characterization of its infection cycle revealed unexpected aspects of its structural organization, and of its entry, replication and egress mechanisms [
4,
5,
6,
7]. SIRV2 replicates fast, has a clear and dramatic effect on the host upon infection, and is therefore an appealing model to study crenarchaeal viruses.
The linear dsDNA genome of SIRV2 (35 kb) carries inverted terminal repeats (ITR) and encodes 54 open reading frames (ORFs) [
8]. SIRV2 infects the thermoacidophilic archaeon
S. islandicus LAL14/1, which was isolated from solfatares in Iceland and grows optimally at 78 °C and a pH of 3 [
9]. It has stiff rod-shaped virions of about 900 nm in length and 23 nm in diameter [
9]. The virions consist of multiple copies of the major capsid protein Gp26 enwrapping the linear dsDNA genome. Interestingly, this genome is organized as A-form DNA inside the viral particle, probably to protect the DNA against the high temperature and low pH of the natural environment of
S. islandicus [
10]. The proteins Gp33 and Gp39 are also part of the SIRV2 virions, although in minor amounts [
11]. At each end of the non-enveloped virions three tail fibers are displayed, which consist of multiple copies of the protein Gp38 and are important for virion attachment to the host cell during the entry process [
7]. The tail fibers bind specifically to pili-like structures of the host and virions travel along them to the cell surface, where they deliver the DNA into the host cytoplasm by an unknown mechanism [
7]. The host genome is then rapidly eliminated and the cell is transformed into an efficient virion-producing factory. SIRV1 is another member of the
Rudiviridae that is closely related to SIRV2. It was isolated in Iceland at a separate location from SIRV2, and infects
S. islandicus KVEM10H3. It has a similar genome organization and morphology as SIRV2 [
9]. The main difference between SIRV1 and SIRV2 is that SIRV1 encodes nine fewer genes, and that it has an unusual genome instability, which is illustrated by the high number of available genetic variants [
8,
12]. Therefore, the more stable SIRV2 is more amenable to virus–host interaction studies.
As a first step during archaeal viral infection, the viral genomes are replicated. The genome organization of
Rudiviridae with their ITRs is reminiscent of that of large cytoplasmic DNA viruses, such as the
Poxviridae [
13]. However, the rudiviruses replicate by a novel mechanism involving a Rep-like protein, Gp16 [
6]. Gp17 and Gp18 were also suggested to play roles in replication [
14]. During replication, head-to-head and tail-to-tail replicative intermediates are formed, which can be resolved by the virus-encoded Holliday junction resolvase Gp35 [
6,
15]. After the SIRV genome has been replicated, new linear virions are formed in the cytoplasm of the host cell, by the packaging of the DNA genome with the coat protein Gp26. Simultaneously, preparations are made for virion release. Multiple heptagonal pyramidal-shaped structures are formed on the cell surface [
4]. These virus-associated pyramids (VAPs) consist of multiple copies of the virus-encoded membrane protein forming Virus-Associated Pyramids (PVAP) (Gp49) and open outwards creating large apertures (~200 nm) through which the virions can egress [
5,
16,
17]. This unique virus egress mechanism was demonstrated to exist only in a small set of crenarchaeal viruses; i.e., SIRV2 and STIV1 (
Sulfolobus turreted icosahedral virus) [
16,
17].
In contrast to most archaeal viruses, quite a number of genes of SIRV2 already have predicted or assigned functions [
3]. Still, the functions of about half of all SIRV2 genes are unknown and await functional characterization to obtain further insights into the SIRV infection cycle. One of these uncharacterized proteins is Gp1, named SIRV2_Gp1 throughout this paper to discriminate from its SIRV1 homolog (SIRV1_Gp1). Previously, this protein was also referred to as ORF83a/ORF83b depending on the genomic location of its encoding gene [
18]. SIRV2_Gp1 (and SIRV1_Gp1) is encoded twice in the viral genome: both genes have identical DNA sequences and are located at each genome terminus [
8,
18]. Transcriptomic analysis of the SIRV2 infectious cycle showed that both gene copies are transcribed at very high levels during the very first stages of infection and that their expression levels remain high throughout the infection cycle [
18,
19].
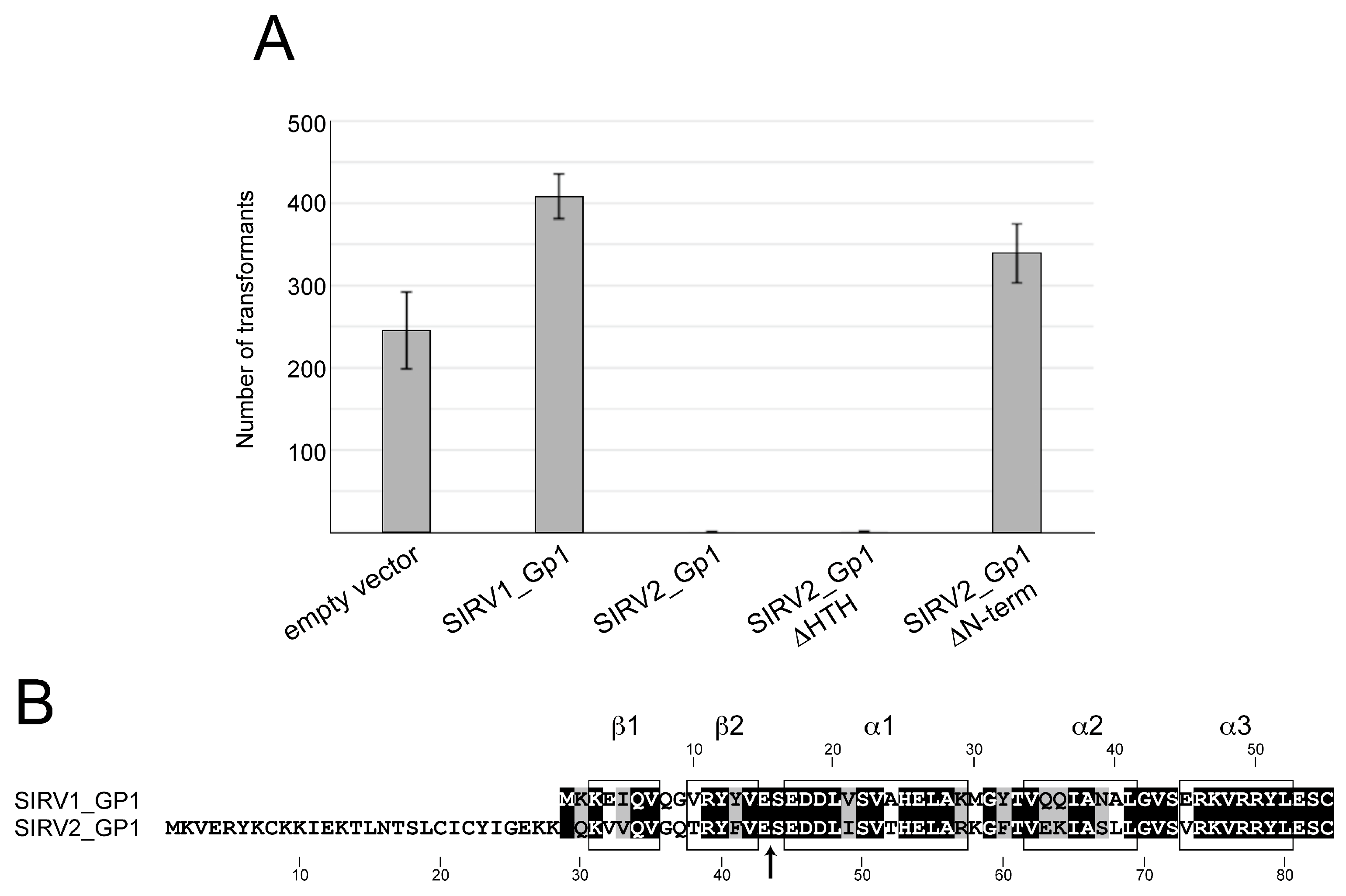
Gene duplication and high expression levels suggest an important function of SIRV2_Gp1 with regards to the infection process. However, its function remains elusive. Given that it is a small 8-kDa protein almost entirely characterized by a helix-turn-helix (HTH) motif, typical of DNA-binding proteins, we aimed to functionally characterize this protein by studying its putative ability to interact with DNA, using electrophoretic mobility shift assays (EMSAs) and atomic force microscopy (AFM). These investigations showed that SIRV2_Gp1 is capable of binding and condensing dsDNA. Furthermore, by using a Sulfolobus acidocaldarius expression system we provided proof that SIRV2_Gp1 is a highly toxic protein although the HTH motif does not seem to contribute to the observed DNA-binding and toxicity characteristics of the protein.
2. Materials and Methods
2.1. Protein Purification
The
SIRV2_gp1 open reading frame and its truncated variant (
SIRV2_gp1 ∆HTH) were amplified with primers 1 and 2 and 1 and 22, respectively (
Table S1) from Integrated DNA Technologies (IDT, Coralville, IA, USA) and were cloned with C-terminal His-tag in pEXP5-CT/TOPO. The plasmids encoding SIRV1_Gp1 and SIRV1_Gp ∆HTH were transformed into
Escherichia coli Rosetta™ (DE3)pLysS Competent Cells (Novagen, Madison, WI USA) and BL21 (DE3) pLysS chemically competent cells, respectively. Cells were grown in LB (Luria–Bertani) medium supplemented with 50 μg/mL ampicillin and grown to an optical density of 600 nm (OD
600) of ~0.4–0.8 at 37 °C. Recombinant protein expression was then induced by the addition of 1 mM isopropyl-β-
d-thiogalactopyranoside (IPTG) and cells were grown for 3 more hours at 37 °C. Cells were pelleted and resuspended in lysis buffer (50 mM Tris pH8 500 mM NaCl, 30 mM imidazole, 1 mg/mL lysozyme, protease inhibitor (Roche Applied Science, Basel, Switzerland). Cells were lysed by sonication, the lysate was cleared by ultracentrifugation and the supernatant was filtered through a 0.22 μm syringe filter and loaded on to a 1 mL Protino
® Ni-NTAcolumn (Machery-Nagel, Bethlehem, PA, USA) equilibrated in buffer A (50 mM Tris pH 8500 mM NaCl, 30 mM imidazole). SIRV2_Gp1 was eluted with a linear gradient from 30 to 500 mM imidazole. Peak fractions were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and the fractions containing the highest amounts of protein were pooled, filtered on a 0.22 μm syringe filter and directly loaded on a HiLoad 16/600 SuperDex 75 pg column (GE Healthcare, Little Chalfont, UK), without prior concentration. The protein was run on the gelfiltration column in buffer C (20 mM MES pH 6.5, 300 mM NaCl, 1 mM DTT, 1 mM EDTA). Purified and concentrated protein samples were flash frozen and stored at −80 °C. The SIRV2_gp1 ∆HTH truncation mutant protein was recombinantly purified following a similar procedure as for the full-length protein with the following change: lysis buffer and Buffer A did not contain imidazole. The
SIRV1_gp1 gene was cloned and the corresponding protein was expressed and purified with immobilized metal affinity chromatography (Ni-IMAC) and gel filtration chromatography as described by Oke et al. [
20]. The crystallization and structure solution of SIRV1_Gp1 have been previously described [
20], and the coordinates are available from the Protein Data Bank (PDB) (identifier [ID] 2X48).
2.2. Electrophoretic Mobility Shift Assays
Different 5′ fluorescein amidite (6-FAM) labeled random 30 bp oligonucleotides were ordered from IDT. Oligos 3–4 (for dsDNA), 5 (for hairpin DNA) and 7–10 (for Holliday junctions) (see
Table S1) were annealed by heating with an excess of unlabeled strands at 90 °C for 2 min and then slowly cooling to room temperature overnight in a heating block. In case of single-stranded (ss) DNA, no prior heating occurred and oligo 3 or 4 were used alone. The assembled substrates were purified by native polyacrylamide (12%) gel electrophoresis with 1× Tris-borate-EDTA (TBE) buffer, followed by band excision, gel extraction and ethanol precipitation before being resuspended in water to a concentration of 1 μM for use in assays. The final concentration in assays was 100 nM. Serial dilutions of purified protein and labeled oligonucleotides were mixed in reaction buffer (50 mM Tris pH 7.5, 5 mM EDTA, 1 mM DTT, 100 μg/mL bovine serum albumin (BSA). After a 20 min incubation at room temperature, samples were mixed in a 2:1 ratio with ficoll, loaded on 8% Tris-Borate-EDTA (TBE) gel and electrophoresed at 180 V during 1 to 2 h. After electrophoresis, the gels were scanned using a Fujifilm FLA-5000 imager at a wavelength of 473 nm.
EMSAs with specific DNA fragments were performed as described previously [
21]. Briefly, different concentrations of SIRV2_Gp1 or SIRV2_Gp1 ∆HTH protein were mixed with 5′-end
32P-labeled probes in presence of an excess of unlabeled salmon sperm DNA (25 ng/μL) in reaction buffer (20 mM Tris pH 8.0, 0.4 mM EDTA, 1 mM MgCl
2, 0.1 mM DTT, 12.5% glycerol, 50 mM NaCl) and incubated for 25 min at 37 °C prior to analysis by native acryalamide gel electrophoresis. The labeled probes are a 236 bp fragment corresponding to the region upstream of the SIRV2_GP1-encoding ORF (prepared with primers ep399 and ep400,
Table S1) and a 173 bp unspecific promoter fragment of
S. acidocaldarius (prepared with primers ep092 and ep093,
Table S1) for SIRV2_Gp1 binding and a 102 bp unspecific fragment of
S. acidocaldarius (prepared with primers LL139 and LL140,
Table S1) for SIRV2_Gp1 ∆HTH binding. Bands were visualized by autoradiography.
EMSAs with plasmid DNA were performed by mixing 100 ng pUC19 DNA (New England Biolabs, Ipswich, MA, USA) with different concentrations of protein in reaction buffer 1 (50 mM Tris pH 7.5, 5 mM EDTA, 1 mM 1,4-Dithiothreitol (DTT), 100 μg/mL BSA) or 2 (20 mM Tris, pH 8.0, 1 mM MgCl, 50 mM NaCl, 0.4 mM EDTA, 0.1 mM DTT, 12.5% glycerol), which gave the same results. After an incubation of 20 min at room temperature, samples were mixed in a 1:5 ratio with 6× DNA loading dye (Thermo Scientific, Waltham, MA, USA) and loaded on an ethidium bromide gel, which was run for 30 min at 100 V after which bands were visualized with an ultraviolet (UV) scanner.
2.3. Cleavage Assays
5′-FAM labeled oligonucleotides (see above) and 5 µM of protein were mixed in reaction buffer (20 mM Tris pH 7.5, 10 mM NaCl, 1 mM DTT, 10 mM MgCl) and incubated during 30 min at 50 °C. One unit of Proteinase K was added, samples were incubated at 37 °C and after 30 min, formamide was added 1:2 to the reaction mixture. Samples were loaded on a 20% Urea TBE gel and run at 22 W at 45 °C for 2–3 h.
2.4. Atomic Force Microscopy
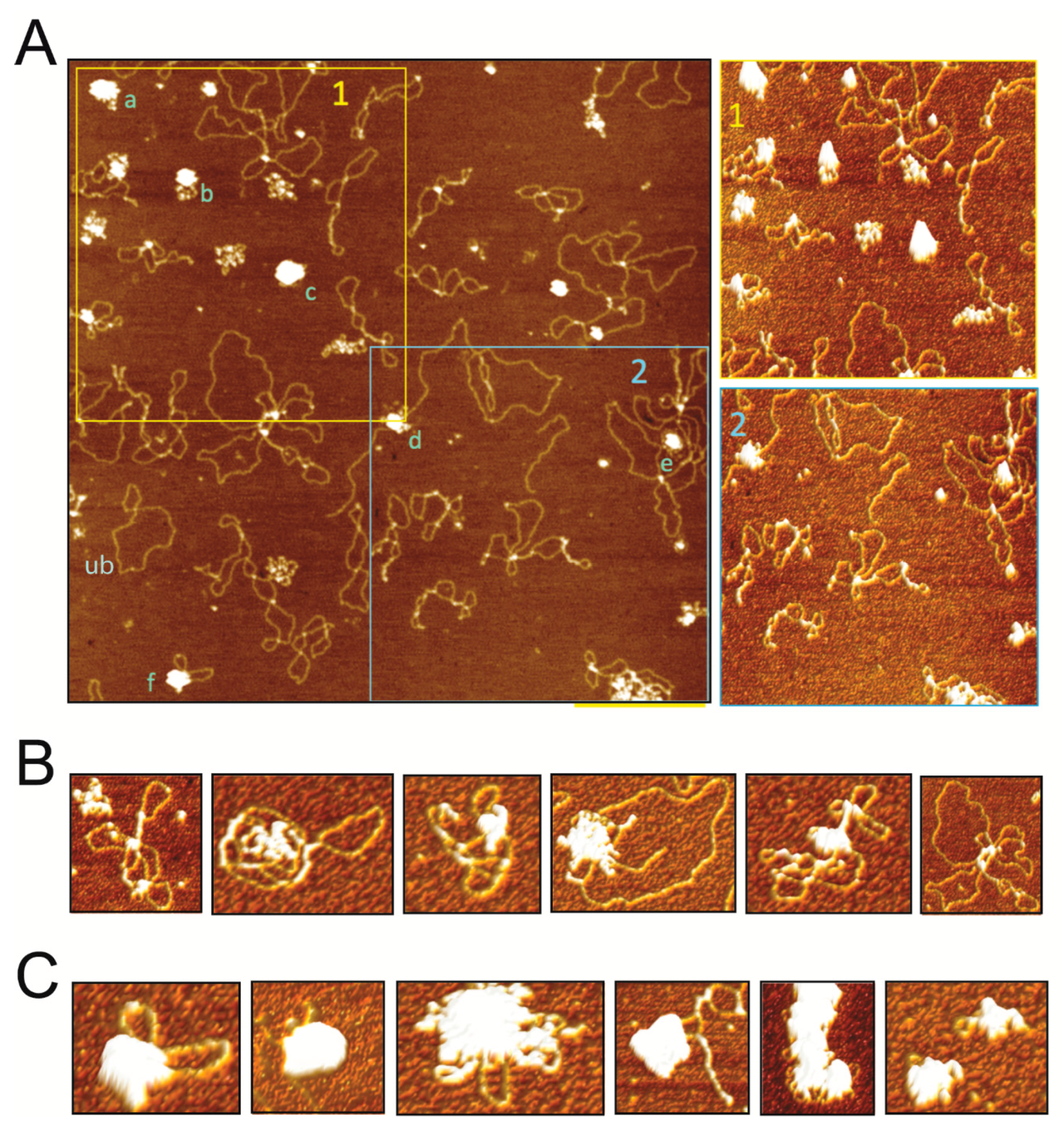
For AFM imaging, protein-DNA binding mixtures containing 50 nM pUC18 plasmid DNA and 15 nM-30 nM SIRV2_Gp1 protein were prepared in adsorption buffer (40 mM HEPES pH 6.9, 10 mM NiCl2) and deposited on freshly cleaved mica. After 5 min incubation, the mica surface was rinsed with deionized ultrapure water and blown dry with a gentle stream of nitrogen. Images were collected with a MultiMode (NanoScope IIIa) AFM (Bruker, Billerica, MA, USA) operated in tapping mode in air using RTESP (Bruker) AFM tips (cantilever length of 115–135 μm, width of 30–40 µm, a nominal spring constant of 20–80 N/m, and resonance frequencies in the range from 264 to 284 kHz). NanoScope Analysis v1.5 software (Bruker) was used to flatten the images, perform cross-section analyses of the complexes, and to make three-dimensional (3D) surface plots of selected complexes with a pitch of 3°.
2.5. Toxicity Assay
The
SIRV1_gp1 and
SIRV2_gp1 genes and two truncation mutants of the
SIRV2_gp1 gene lacking 84 bp on the 5′ end (SIRV2_Gp1 ∆N-term) or 117 bp on the 3′ end (SIRV2_Gp1 ∆HTH), were amplified from viral genomic DNA with primers 16 + 17, 1 + 2, 18 + 19 and 20 + 21 respectively (
Table S1). The genes were cloned in a pENTR™/SD/D-TOPO
® vector according to manufacturer’s protocol and transformed to One Shot
® TOP10 Chemically Competent
E. coli (Thermo Scientific). Next the genes were introduced via Gateway
® (Thermo Scientific) cloning in the maltose inducible expression plasmid for
S. acidocaldarius, pSVA1551 [
22]. pSVA1551 harbors the
pyrEF-encoded proteins, which allow for selection on uracil-free medium when expressed in
S. acidocaldarius MW001 (Δ
pyrEF). Plasmids were methylated in
E. coli ER1828 and 150 ng was transformed to the
S. acidocaldarius MW001 via electroporation as described earlier [
23]. The cells were plated on selective Brock Gelrite plates lacking uracil, which were supplemented with 0.2% dextrin and NZ amine. Colonies were grown at 75 °C during 6 days. The experiment was performed independently three times using quadruplicates of each strain.
4. Discussion
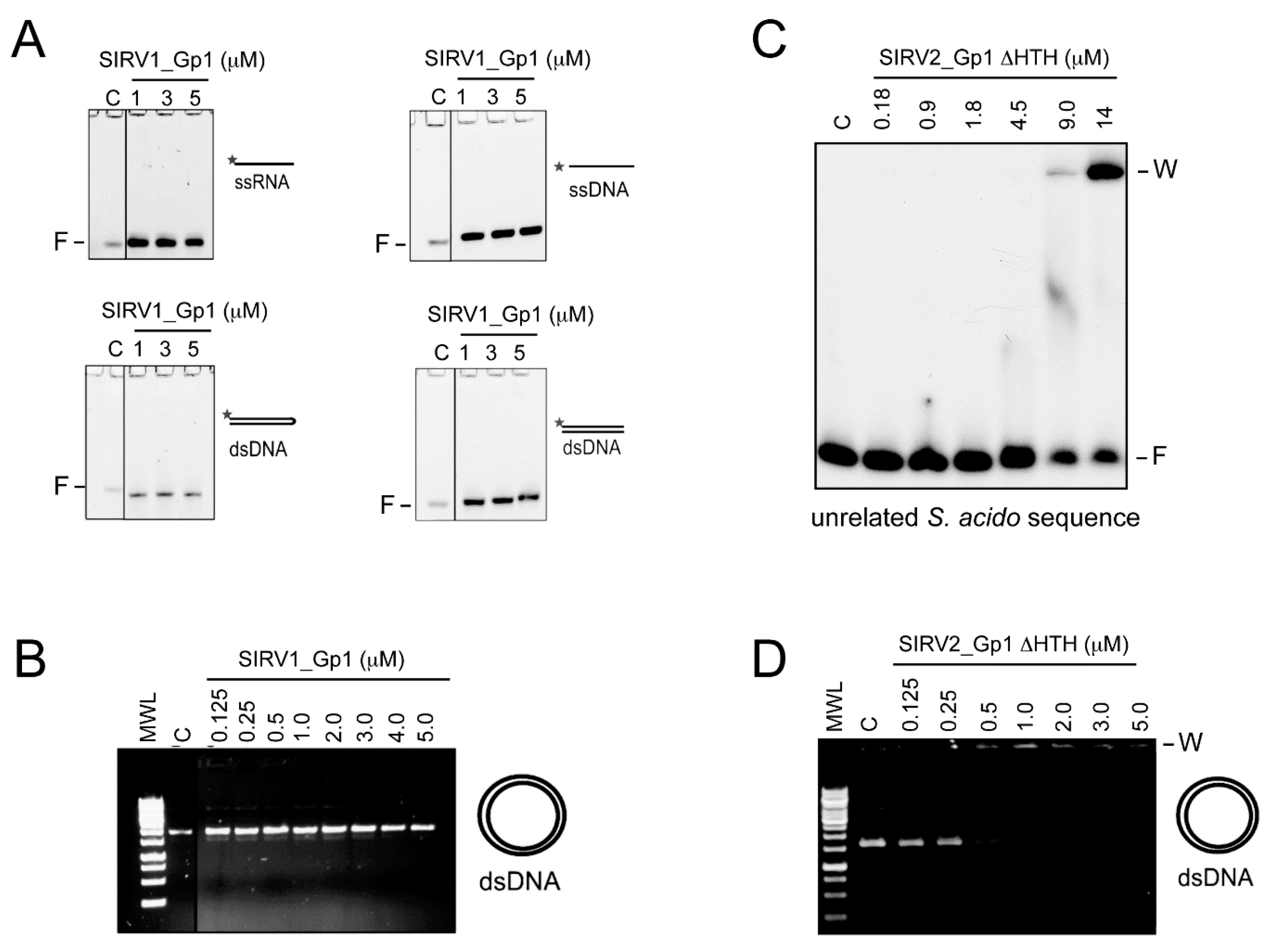
In this study, we demonstrated that the Rudiviral protein SIRV2_Gp1 binds several nucleic acid species with a preference for dsDNA. This binding appears to lack sequence specificity given the observation that SIRV2_Gp1 significantly retards migration of short randomized or large plasmid DNA probes (
Figure 1 and
Figure 4). However, we cannot exclude the possibility that SIRV2_Gp1 might bind a yet unidentified sequence with higher specificity. Furthermore, study of the architecture of SIRV2_Gp1 nucleoprotein complexes revealed protein-induced aggregation zones in dense complexes. Employing the
S. acidocaldarius genetic system, we further showed that SIRV2_Gp1 is toxic to
Sulfolobus cells and that this toxicity is caused by a lysine-rich N-terminal extension, which also mediates DNA binding and in which the typical HTH motif does not seem to be involved. The shorter SIRV1 version of Gp1 was not toxic to
Sulfolobus cells and EMSAs indicated that this protein is unable to interact with DNA.
Upon aligning
SIRV1_
gp1 and
SIRV2_
gp1 DNA sequences (
Figure S3), the correctness of ORF annotation could be questioned. To analyze the transcriptional structure of the
SIRV1_
gp1 gene, we aimed at analyzing transcriptome data. While the many repeats encoded in this genome region have hampered a Northern blot expression analysis of
SIRV1_
gp1 [
27], the stable replication and high virus production of SIRV2 have allowed for a recent RNA-seq analysis [
18]. In this study, transcription levels of
SIRV2_
gp1 were quantified at several time points during infection. Based on these data, it appears that the
SIRV2_
gp1 gene is characterized by a transcriptional dynamic resulting in two alternative transcripts that are translated from different start codons yielding the full-length and truncated SIRV2_Gp1 protein, respectively. At early stages of infection, hardly any reads covering the 5′-region of
SIRV2_
gp1 were detected, suggesting that, at that time point, possibly only a short version of
gp1, encoding the SIRV1_Gp1 homolog lacking the N-terminal extension, is expressed [
18]. However, later during SIRV2 infection the long version of the
gp1 gene appears to be transcribed, although the coverage of the 5′-region is still considerably lower than the 3′-region [
18]. Therefore, the shorter 55 amino-acid version of SIRV2_Gp1 might be the dominant species during SIRV2 infection, while at later stages the longer 83 amino-acid protein might become relevant. The massive DNA condensation caused by the 83 amino-acid version and its apparent toxicity might be compatible to a role in elimination of the host defense system. The absence of the longer Gp1 version in SIRV1, and the subsequent absence of DNA condensation, wrapping activity and toxicity, seems in concert with the observed mild and partially defective progression of infection by SIRV1.
The observation of the N-terminal extension of the full-length SIRV2_Gp1 protein mediating host toxicity by DNA condensation does not inform us about the putative function of the truncated version expressed during early stages of infection and of the corresponding SIRV1_Gp1 ortholog. Previously, it was shown that the SIRV2_Gp1 protein interacts with a Holliday junction resolvase (encoded by ORF121 in SIRV2) [
18] and the PCNA3 (proliferating cell nuclear antigen) subunit of the
Sulfolobus sliding clamp, a processivity factor of archaeal DNA polymerase [
28]. Based on this observation, SIRV2_Gp1 was hypothesized to be implicated in the initiation of viral genome replication and/or the resolution of viral replicative intermediates [
28,
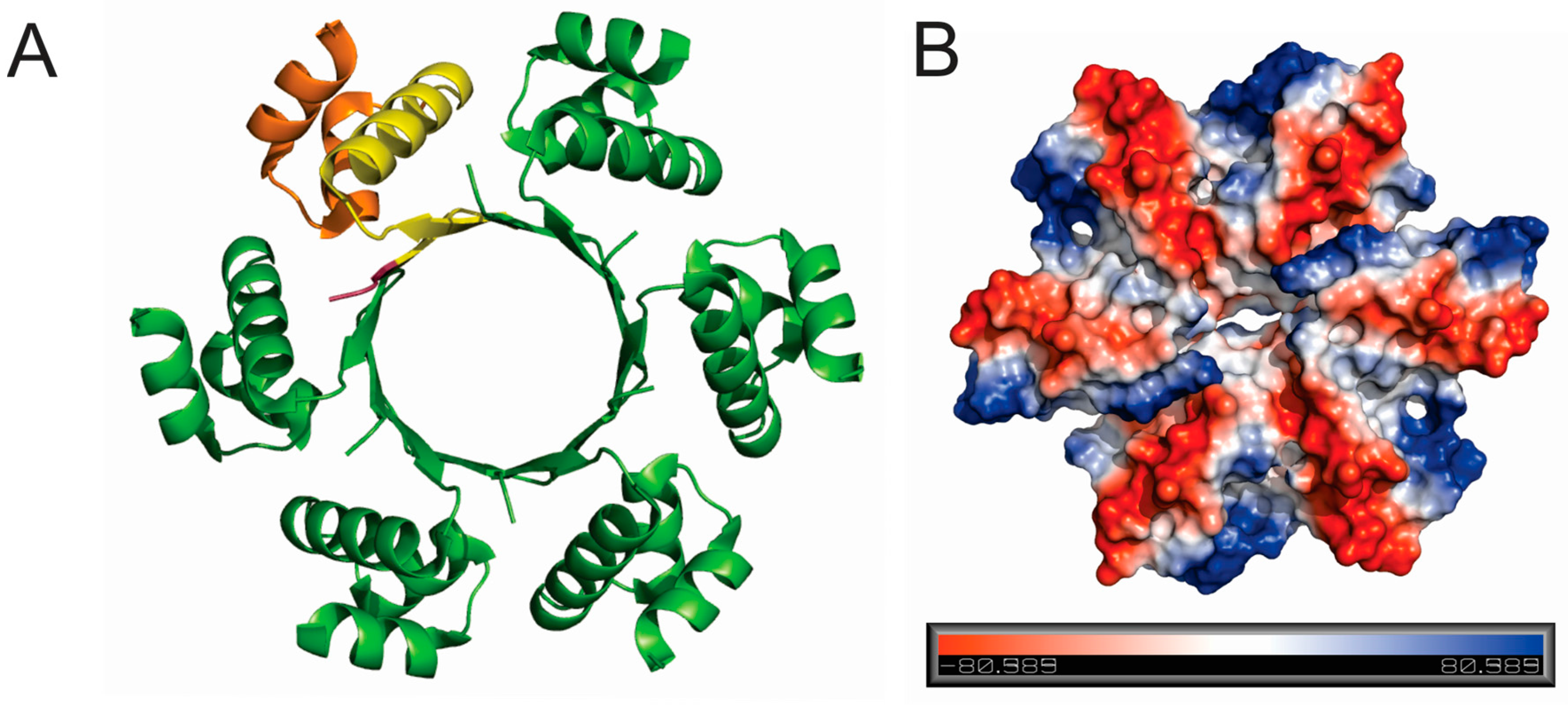
29]. It could thus be envisaged that SIRV2_Gp1 has a dual function, depending on its translational length, and that it assists in viral replication during early stages of the infection while condensing the host genome during later stages. The lack of observed nucleic acid-binding activity in vitro for SIRV1_Gp1, despite the presence of the HTH motif, was unexpected given the unequivocal implication of this motif in DNA binding. Possibly, the assembly into a hexameric ring in vitro (
Figure 5) prevents interaction with DNA because of a suboptimal relative positioning with respect to consecutive helical turns of a DNA molecule. In vivo, a heterooligomeric assembly of SIRV1_Gp1 (or the truncated SIRV2_Gp1 protein) and the resolvase might harbour DNA-binding activity.
The massive DNA wrapping and condensation activity as observed for SIRV2_Gp1 might be employed as an inducible toxic peptide in a biotechnological setting for containment of the spread of genetically modified organisms or as a viral weapon for killing pathogenic bacteria. In addition to this biotechnological relevance, our findings contribute to the understanding of the Rudiviral infection cycle and pave the way for further study of archaeal viruses in general.



 and
and 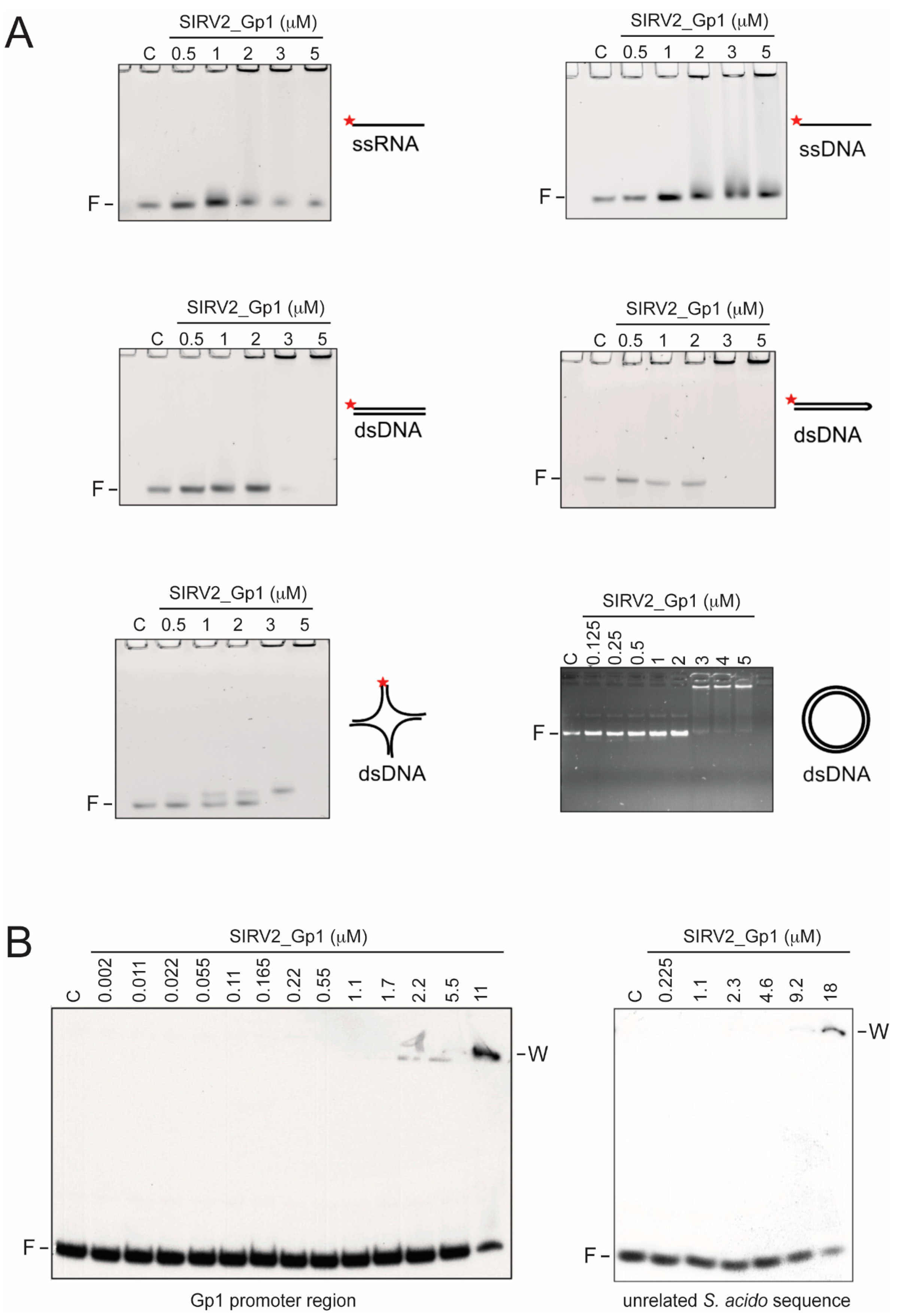{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




