
The Interaction between Nidovirales and Autophagy Components



Abstract
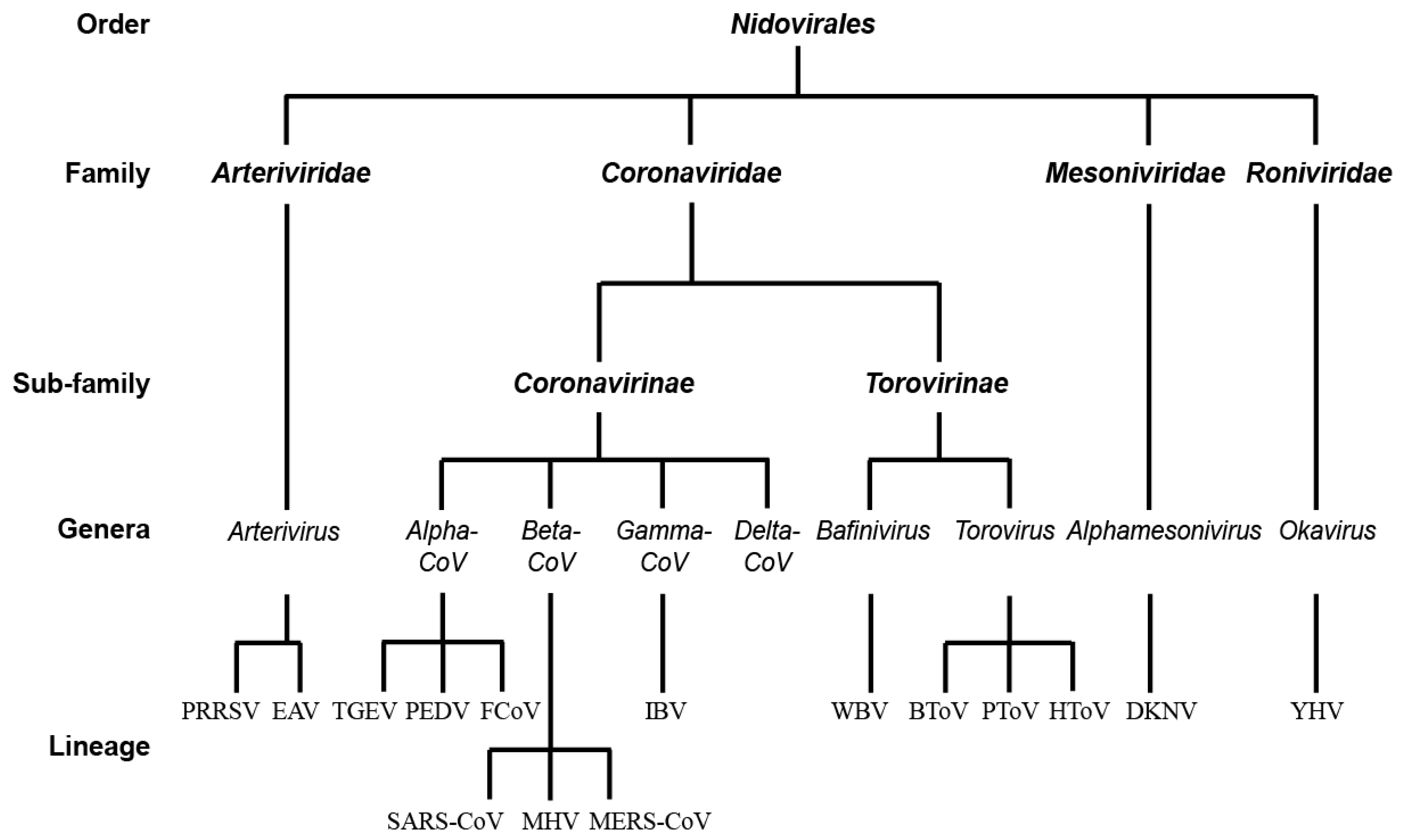
:1. The Order of Nidovirales
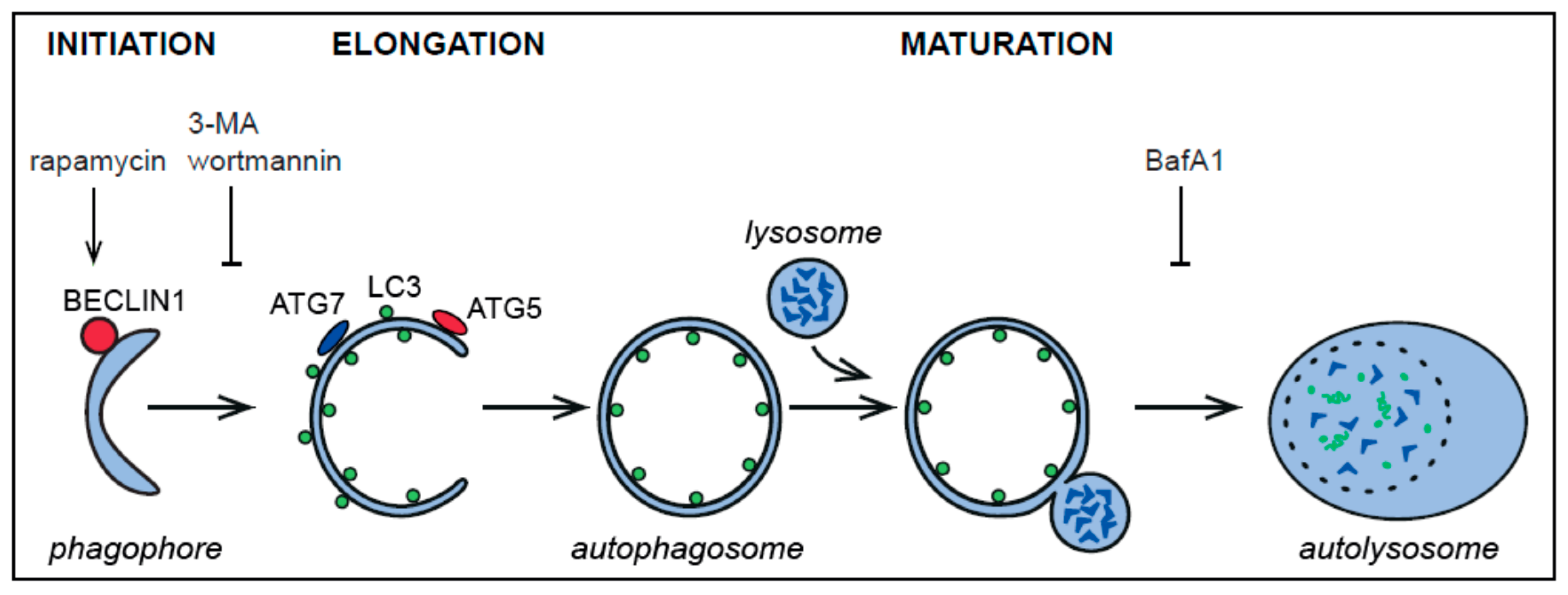
2. Autophagy and the Autophagy-Related Proteins
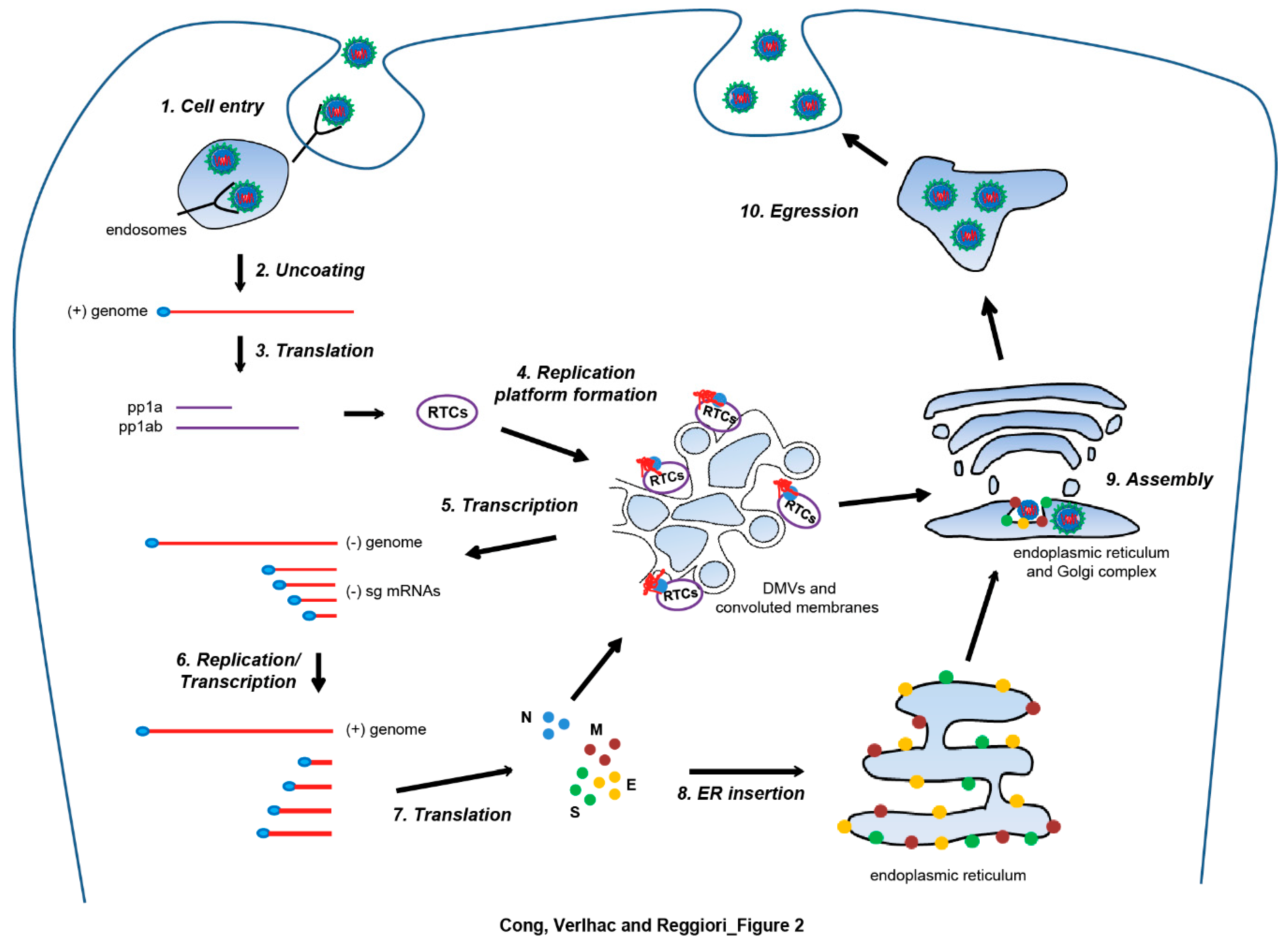
3. Nidovirales and Autophagy
3.1. Arteriviruses and Autophagy
3.2. Alphacoronaviruses and Autophagy
3.3. Betacoronaviruses and Autophagy
3.4. Gammacoronavirus and Autophagy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Revision of the taxonomy of the Coronavirus, Torovirus and Arterivirus genera. Arch Virol. 1994, 135, 227–237.
- Lai, M.M.; Cavanagh, D. The molecular biology of coronaviruses. Adv. Virus Res. 1997, 48, 1–100. [Google Scholar] [PubMed]
- De Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.M.; Talbot, P.J. Coronaviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier Academic Press: San Diego, CA, USA, 2012; pp. 774–796. [Google Scholar]
- Pasternak, A.O.; Spaan, W.J.; Snijder, E.J. Nidovirus transcription: How to make sense...? J. Gen. Virol. 2006, 87, 1403–1421. [Google Scholar] [CrossRef] [PubMed]
- Siddell, S.G. The Coronaviridae. In The Coronaviridae; Springer: New York, NY, USA, 1995; pp. 1–10. [Google Scholar]
- McCluskey, B.J.; Haley, C.; Rovira, A.; Main, R.; Zhang, Y.; Barder, S. Retrospective testing and case series study of porcine delta coronavirus in US swine herds. Prev. Vet. Med. 2016, 123, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Ren, X. Coronavirus entry and release in polarized epithelial cells: A review. Rev. Med. Virol. 2014, 24, 308–315. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.A.F.; Horzinek, M.C.; Rottier, P.J.M.; De Groot, R.J. The Genome Organization of the Nidovirales: Similarities and Differences Between Arteri-, Toro-, and Coronaviruses; Seminars in VIROLOGY; Elsevier: Lincoln, UK, 1997; pp. 33–47. [Google Scholar]
- Cong, Y.; Zarlenga, D.S.; Richt, J.A.; Wang, X.; Wang, Y.; Suo, S.; Wang, J.; Ren, Y.; Ren, X. Evolution and homologous recombination of the hemagglutinin-esterase gene sequences from porcine torovirus. Virus Genes 2013, 47, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Vasilakis, N.; Guzman, H.; Firth, C.; Forrester, N.L.; Widen, S.G.; Wood, T.G.; Rossi, S.L.; Ghedin, E.; Popov, V.; Blasdell, K.R.; et al. Mesoniviruses are mosquito-specific viruses with extensive geographic distribution and host range. Virol. J. 2014, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Zirkel, F.; Roth, H.; Kurth, A.; Drosten, C.; Ziebuhr, J.; Junglen, S. Identification and characterization of genetically divergent members of the newly established family Mesoniviridae. J. Virol. 2013, 87, 6346–6358. [Google Scholar] [CrossRef] [PubMed]
- Zirkel, F.; Kurth, A.; Quan, P.L.; Briese, T.; Ellerbrok, H.; Pauli, G.; Leendertz, F.H.; Lipkin, W.I.; Ziebuhr, J.; Drosten, C.; et al. An insect nidovirus emerging from a primary tropical rainforest. mBio 2011, 2, e00077-11. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Liu, S.; Zhu, L.; Wan, X.; Liu, Q.; Qiu, L.; Zou, P.; Zhang, Q.; Huang, J. Complete genome sequence of an isolate of a novel genotype of yellow head virus from Fenneropenaeus chinensis indigenous in China. Arch Virol. 2017, 162, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Prather, R.S.; Whitworth, K.M.; Schommer, S.K.; Wells, K.D. Genetic engineering alveolar macrophages for host resistance to PRRSV. Vet. Microbiol. 2017, 16, S0378–1135. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.J.; Kliebenstein, J.B.; Johnson, C.D.; Mabry, J.W.; Bush, E.J.; Seitzinger, A.H.; Green, A.L.; Zimmerman, J.J. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J. Am. Vet. Med. Assoc. 2005, 227, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, S.; Sola, I.; Alonso, S.; Enjuanes, L. Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J. Virol. 2004, 78, 980–994. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; Rottier, P.J.M. Nidovirus entry into cells. In Nidoviruses; American Society of Microbiology: Washington, DC, USA, 2008; pp. 157–178. [Google Scholar]
- Ziebuhr, J.; Siddell, S. Nidoviruses. eLS 2008. [Google Scholar] [CrossRef]
- Yokomori, K.; Banner, L.R.; Lai, M.M.C. Heterogeneity of gene expression of the hemagglutinin-esterase (HE) protein of murine coronaviruses. Virology 1991, 183, 647–657. [Google Scholar] [CrossRef]
- Ulferts, R.; Ziebuhr, J. Nidovirus ribonucleases: Structures and functions in viral replication. RNA Biol. 2011, 8, 295–304. [Google Scholar] [CrossRef] [PubMed]
- De Haan, C.A.M.; Reggiori, F. Are nidoviruses hijacking the autophagy machinery? Autophagy 2008, 4, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Enjuanes, L.; Gorbalenya, A.E.; De Groot, R.J.; Cowley, J.A.; Ziebuhr, J.; Snijder, E.J. Nidovirales; Elsevier: Amsterdam, The Netherlands, 2000; pp. 785–795. [Google Scholar]
- Lavi, E.; Weiss, S.R.; Hingley, S.T. The Nidoviruses: (Coronaviruses and Arteriviruses); Springer Science & Business Media: Berlin, Germany, 2012; Volume 494, pp. 609–698. [Google Scholar]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, M.E.; Tavernarakis, N. Autophagy and the endo/exosomal pathways in health and disease. Biotechnol. J. 2017, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Lippai, M.; Szatmari, Z. Autophagy-from molecular mechanisms to clinical relevance. Cell Biol. Toxicol. 2017, 33, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy in leukocytes and other cells: Mechanisms, subsystem organization, selectivity, and links to innate immunity. J. Leukoc. Biol. 2016, 100, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Petibone, D.M.; Majeed, W.; Casciano, D.A. Autophagy function and its relationship to pathology, clinical applications, drug metabolism and toxicity. J. Appl. Toxicol. 2017, 37, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, L.E.; Williamson, L.E.; Chan, E.Y. Advances in Autophagy Regulatory Mechanisms. Cells 2016, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Munz, C. Autophagy and Mammalian Viruses: Roles in Immune Response, Viral Replication, and Beyond. Adv. Virus Res. 2016, 95, 149–195. [Google Scholar] [PubMed]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. The autophagosome is overrated! Autophagy 2011, 7, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Munson, M.J.; Ganley, I.G. MTOR, PIK3C3, and autophagy: Signaling the beginning from the end. Autophagy 2015, 11, 2375–2376. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Joachim, J.; Tooze, S.A. Autophagosome formation—The role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin. Cancer Biol. 2013, 23, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Tooze, S.A.; Reggiori, F. The puzzling origin of the autophagosomal membrane. F1000 Biol. Rep. 2011, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Karanasios, E.; Stapleton, E.; Manifava, M.; Kaizuka, T.; Mizushima, N.; Walker, S.A.; Ktistakis, N.T. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J. Cell Sci. 2013, 126, 5224–5238. [Google Scholar] [CrossRef] [PubMed]
- Koyama-Honda, I.; Itakura, E.; Fujiwara, T.K.; Mizushima, N. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 2013, 9, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Bestebroer, J.; V’Kovski, P.; Mauthe, M.; Reggiori, F. Hidden behind autophagy: The unconventional roles of ATG proteins. Traffic 2013, 14, 1029–1041. [Google Scholar] [CrossRef]
- Subramani, S.; Malhotra, V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013, 14, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Reggiori, F. ATG proteins: Are we always looking at autophagy? Autophagy 2016, 12, 2502–2503. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Reggiori, F. Using microbes as a key tool to unravel the mechanism of autophagy and the functions of the ATG proteins. Microb. Cell 2016, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kimmey, J.M.; Huynh, J.P.; Weiss, L.A.; Park, S.; Kambal, A.; Debnath, J.; Virgin, H.W.; Stallings, C.L. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 2015, 528, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Langereis, M.; Jung, J. An siRNA screen for ATG protein depletion reveals the extent of the unconventional functions of the autophagy proteome in virus replication. J. Cell Biol. 2016, 214, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Cali, T.; Galli, C.; Olivari, S.; Molinari, M. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys. Res. Commun. 2008, 371, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Le Fourn, V.; Gaplovska-Kysela, K.; Guhl, B.; Santimaria, R.; Zuber, C.; Roth, J. Basal autophagy is involved in the degradation of the ERAD component EDEM1. Cell Mol. Life Sci. 2009, 66, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Zuber, C.; Cormier, J.H.; Guhl, B.; Santimaria, R.; Hebert, D.N.; Roth, J. EDEM1 reveals a quality control vesicular transport pathway out of the endoplasmic reticulum not involving the COPII exit sites. Proc. Natl. Acad. Sci. USA 2007, 104, 4407–4412. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Fang, L.; Wang, D.; Wang, S.; Li, P.; Li, M.; Luo, R.; Chen, H.; Xiao, S. Induction of autophagy enhances porcine reproductive and respiratory syndrome virus replication. Virus Res. 2012, 163, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Pujhari, S.; Kryworuchko, M.; Zakhartchouk, A.N. Role of phosphatidylinositol-3-kinase (PI3K) and the mammalian target of rapamycin (mTOR) signalling pathways in porcine reproductive and respiratory syndrome virus (PRRSV) replication. Virus Res. 2014, 194, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Qin, Y.; Zhou, L.; Kou, Q.; Guo, X.; Ge, X.; Yang, H.; Hu, H. Autophagy sustains the replication of porcine reproductive and respiratory virus in host cells. Virology 2012, 429, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.X.; Huang, L.; Wang, R.; Yu, Y.L.; Li, C.; Li, P.P.; Hu, X.C.; Hao, H.P.; Ishag, H.A.; Mao, X. Porcine reproductive and respiratory syndrome virus induces autophagy to promote virus replication. Autophagy 2012, 8, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yu, Y.; Tu, Y.; Tong, J.; Liu, Y.; Zhang, C.; Chang, Y.; Wang, S.; Jiang, C.; Zhou, E.M.; et al. Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus Infection Induced Apoptosis and Autophagy in Thymi of Infected Piglets. PLoS ONE 2015, 10, e0128292. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, A.; Wang, J.; Zhang, S. Interplay of autophagy and apoptosis during PRRSV infection of Marc145 cell. Infect. Genet. Evol. 2016, 39, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.W.; van der Meer, Y.; Roos, N.; Snijder, E.J. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 1999, 73, 2016–2026. [Google Scholar] [PubMed]
- Zhou, A.; Li, S.; Khan, F.A.; Zhang, S. Autophagy postpones apoptotic cell death in PRRSV infection through Bad-Beclin1 interaction. Virulence 2016, 7, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.N.; Denison, M.R.; Virgin, H.W. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007, 3, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Cali, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Monastyrska, I.; Ulasli, M.; Rottier, P.J.; Guan, J.L.; Reggiori, F.; de Haan, C.A. An autophagy-independent role for LC3 in equine arteritis virus replication. Autophagy 2013, 9, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, W.L.; Lager, K.M.; Vorwald, A.C. Clinical consequences of exposing pregnant gilts to strains of porcine reproductive and respiratory syndrome (PRRS) virus isolated from field cases of “atypical” PRRS. Am. J. Vet. Res. 1998, 59, 1540–1544. [Google Scholar] [PubMed]
- Han, J.; Wang, Y.; Faaberg, K.S. Complete genome analysis of RFLP 184 isolates of porcine reproductive and respiratory syndrome virus. Virus Res. 2006, 122, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Glaser, A.L.; de Vries, A.A.; Rottier, P.J.; Horzinek, M.C.; Colenbrander, B. Equine arteritis virus: A review of clinical features and management aspects. Vet. Q. 1996, 18, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Yin, J.; Li, X.; Yang, W.; Li, G.; Ren, X. Production and characterization of a monoclonal antibody against spike protein of transmissible gastroenteritis virus. Hybridoma 2010, 29, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Park, B. Porcine epidemic diarrhoea virus: A comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes 2012, 44, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoek, L.; Pyrc, K.; Berkhout, B. Human coronavirus NL63, a new respiratory virus. FEMS Microbiol. Rev. 2006, 30, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Rasool, S.; Fielding, B.C. Understanding Human Coronavirus HCoV-NL63. Open Virol. J. 2010, 4, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Shi, H.; Guo, D.; Chen, J.; Shi, D.; Zhu, Q.; Zhang, X.; Feng, L. Analysis of protein expression changes of the Vero E6 cells infected with classic PEDV strain CV777 by using quantitative proteomic technique. J. Virol. Methods 2015, 218, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, M.; Zhang, X.; Tan, X.; Guo, H.; Zeng, W.; Yan, G.; Memon, A.M.; Li, Z.; Zhu, Y.; et al. Porcine Epidemic Diarrhea Virus Induces Autophagy to Benefit Its Replication. Viruses 2017, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yu, H.; Gu, W.; Luo, X.; Li, R.; Zhang, J.; Xu, Y.; Yang, L.; Shen, N.; Feng, L.; Wang, Y. Autophagy Negatively Regulates Transmissible Gastroenteritis Virus Replication. Sci. Rep. 2016, 6, 23864. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Mou, C.; Yang, X.; Lin, J.; Yang, Q. Mitophagy in TGEV infection counteracts oxidative stress and apoptosis. Oncotarget 2016, 7, 27122–27141. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef] [PubMed]
- Prentice, E.; McAuliffe, J.; Lu, X.; Subbarao, K.; Denison, M.R. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J. Virol. 2004, 78, 9977–9986. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; van der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Oostra, M.; te Lintelo, E.G.; Deijs, M.; Verheije, M.H.; Rottier, P.J.; de Haan, C.A. Localization and membrane topology of coronavirus nonstructural protein 4: Involvement of the early secretory pathway in replication. J. Virol. 2007, 81, 12323–12336. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Swett-Tapia, C.; van den Worm, S.H.; Te Velthuis, A.J.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J.; Kikkert, M. Integrity of the early secretory pathway promotes, but is not required for, severe acute respiratory syndrome coronavirus RNA synthesis and virus-induced remodeling of endoplasmic reticulum membranes. J. Virol. 2010, 84, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, R.; Molinari, M. ERAD and ERAD tuning: Disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell Biol. 2011, 23, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, G.A.; van de Nes, P.S.; Bredenbeek, P.J.; Spaan, W.J. The coronavirus spike protein induces endoplasmic reticulum stress and upregulation of intracellular chemokine mRNA concentrations. J. Virol. 2007, 81, 10981–10990. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Alvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, K.; Xing, Y.; Tu, J.; Yang, X.; Zhao, Q.; Li, K.; Chen, Z. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell. 2014, 5, 912–927. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.C.; Devenish, R.J. LC3-Associated Phagocytosis (LAP): Connections with Host Autophagy. Cells 2012, 1, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Farina, A.; Gonnella, R.; Lotti, L.V.; Faggioni, A.; Cirone, M. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 2014, 88, 12715–12726. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.X.; Randall, G. Manipulation or capitulation: Virus interactions with autophagy. Microbes Infect. 2012, 14, 126–139. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Virus | Autophagy Role during Infection | Strategy Used to Modulate Autophagy | Infected Cell/Organ | References |
|---|---|---|---|---|
| TGEV | Antiviral | rapamycin, wortmannin, ATG5/ATG7/LC3 knockdown | Porcine ST cells | [54] |
| Proviral | rapamycin, 3-MA, ATG5 knockdown | Porcine IPEC-J2 cells | [55] | |
| PEDV | Proviral | rapamycin, 3-MA, ATG5/BECLIN1 knockdown | Simian Vero E6 cells | [56] |
| PRSSV | Proviral | rapamycin, 3-MA, BafA1, ATG7/BECLIN1 knockdown | Piglet thymus, simian Marc145 cells | [57,58,59,60,61,62,63,64] |
| MHV | None (LC3 unconventional use) | ATG5/ATG7 knockout, LC3 knockdown | MEFs, human HeLa and HEK293 cells | [65,66,67] |
| EAV | None (LC3 unconventional use) | ATG7 knockout, LC3 knockdown | MEFs, simian Vero E6 cells | [68] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cong, Y.; Verlhac, P.; Reggiori, F. The Interaction between Nidovirales and Autophagy Components. Viruses 2017, 9, 182. https://doi.org/10.3390/v9070182
Cong Y, Verlhac P, Reggiori F. The Interaction between Nidovirales and Autophagy Components. Viruses. 2017; 9(7):182. https://doi.org/10.3390/v9070182
Chicago/Turabian StyleCong, Yingying, Pauline Verlhac, and Fulvio Reggiori. 2017. "The Interaction between Nidovirales and Autophagy Components" Viruses 9, no. 7: 182. https://doi.org/10.3390/v9070182
APA StyleCong, Y., Verlhac, P., & Reggiori, F. (2017). The Interaction between Nidovirales and Autophagy Components. Viruses, 9(7), 182. https://doi.org/10.3390/v9070182