Importance of Autophagy in Mediating Human Immunodeficiency Virus (HIV) and Morphine-Induced Metabolic Dysfunction and Inflammation in Human Astrocytes

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
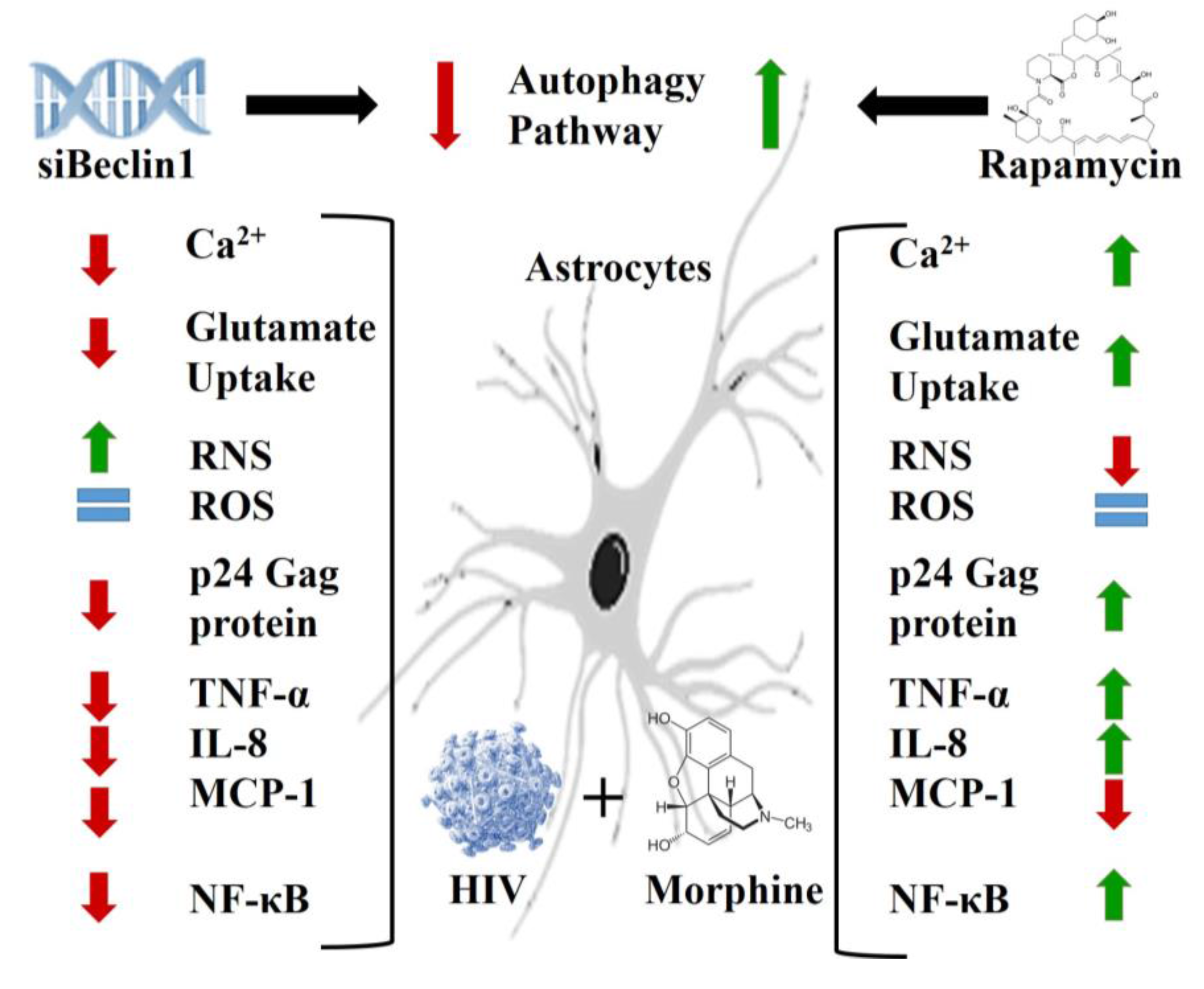{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. HIV Infection, Treatments and Transfection of Human Astrocytes
2.2. Assessment of Intracellular Calcium ([Ca2+]i)
2.3. Glutamate Uptake Measurements
2.4. Reactive Oxygen Species Assay
2.5. Nitric Oxide Assay
2.6. ELISA
2.7. Immunoblotting
2.8. Statistical Analysis
3. Results
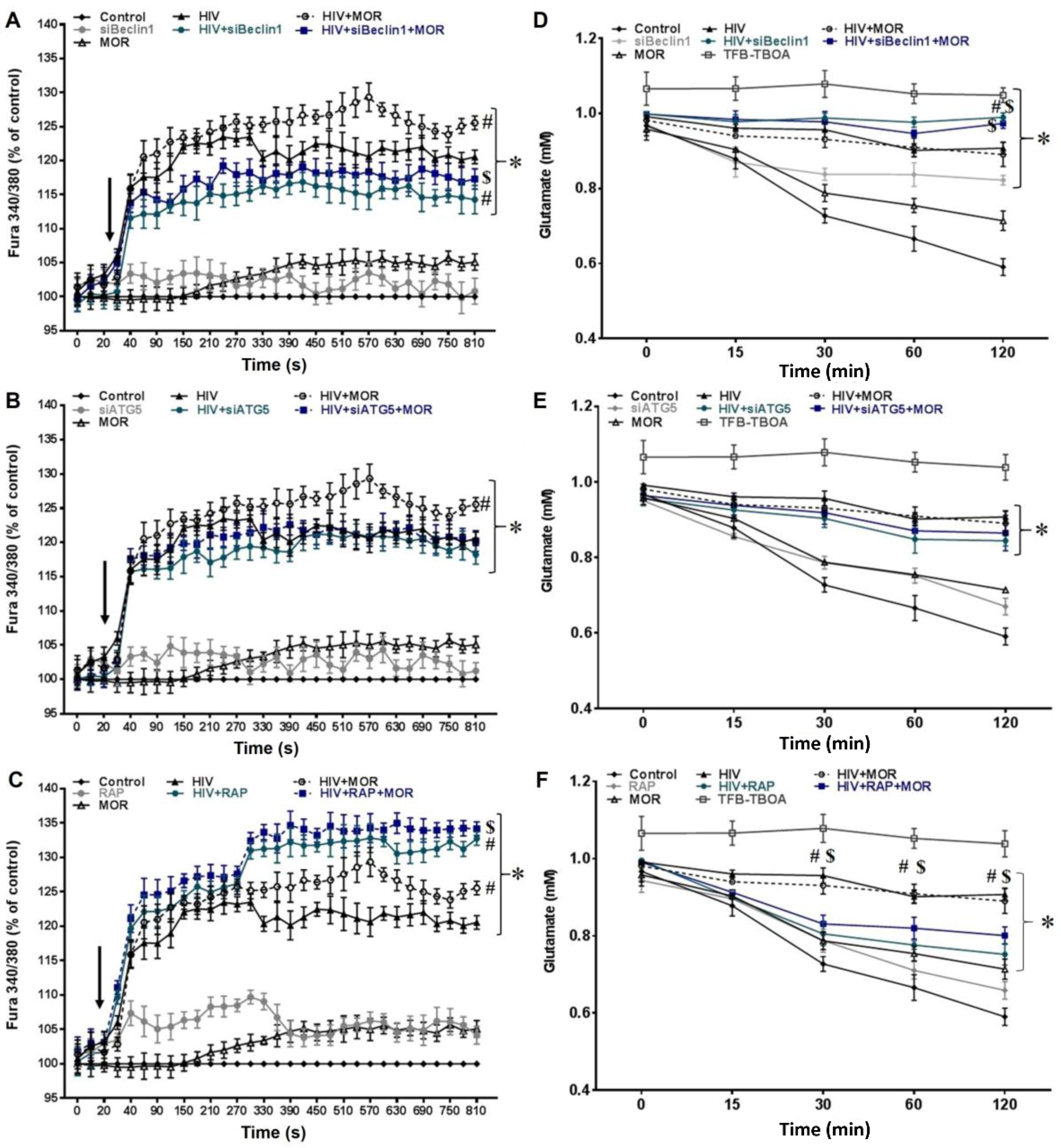
3.1. Role of Autophagy in Mediating HIV and Morphine-Induced Release of Intracellular Calcium ([Ca2+]i) and Glutamate Uptake in Astrocytes.
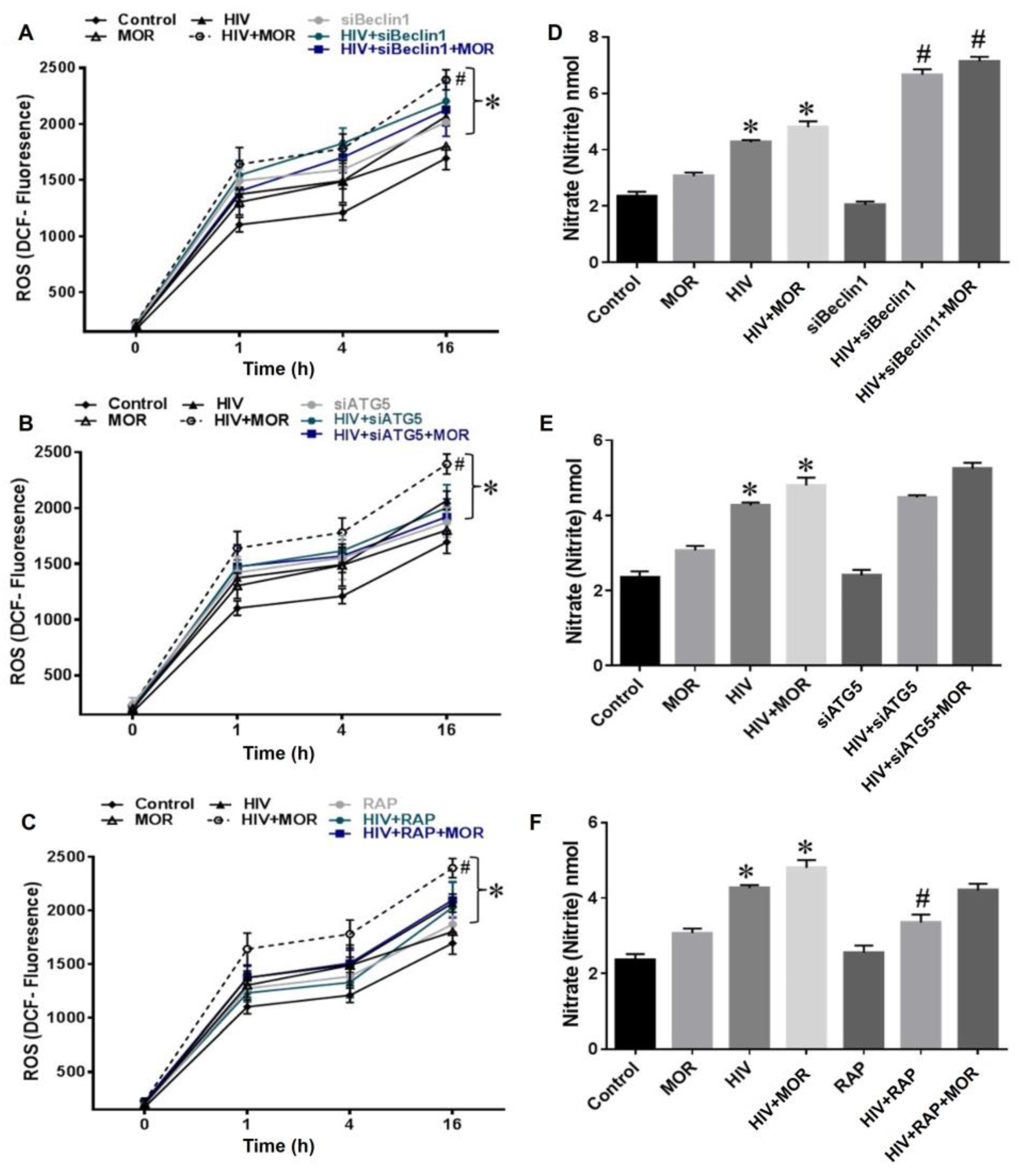
3.2. Role of Autophagy in Mediating HIV and Morphine-Induced Release of Oxyradicals in Astrocytes
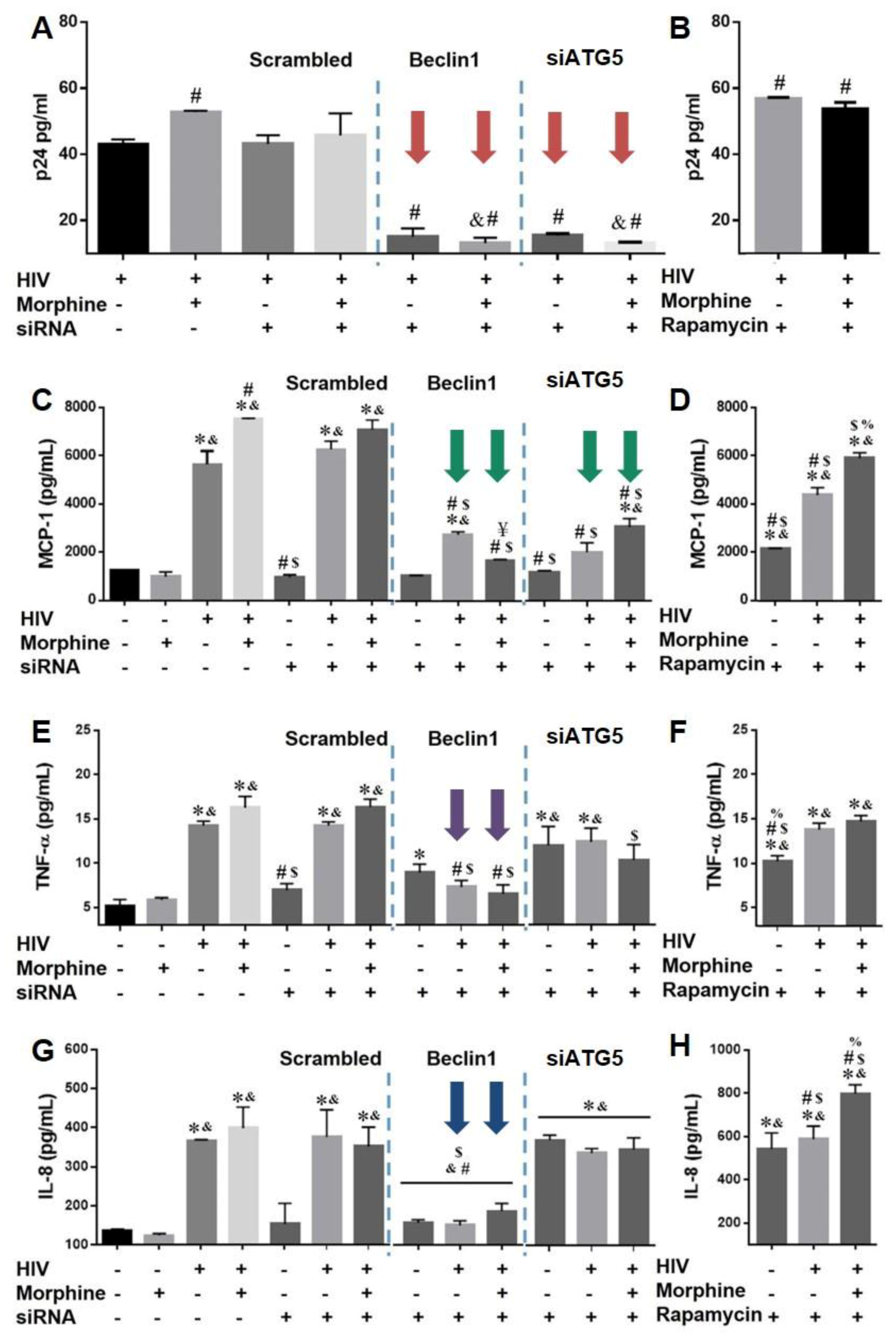
3.3. Role of Autophagy in Mediating HIV Replication and HIV and Morphine-Induced Inflammation in Astrocytes
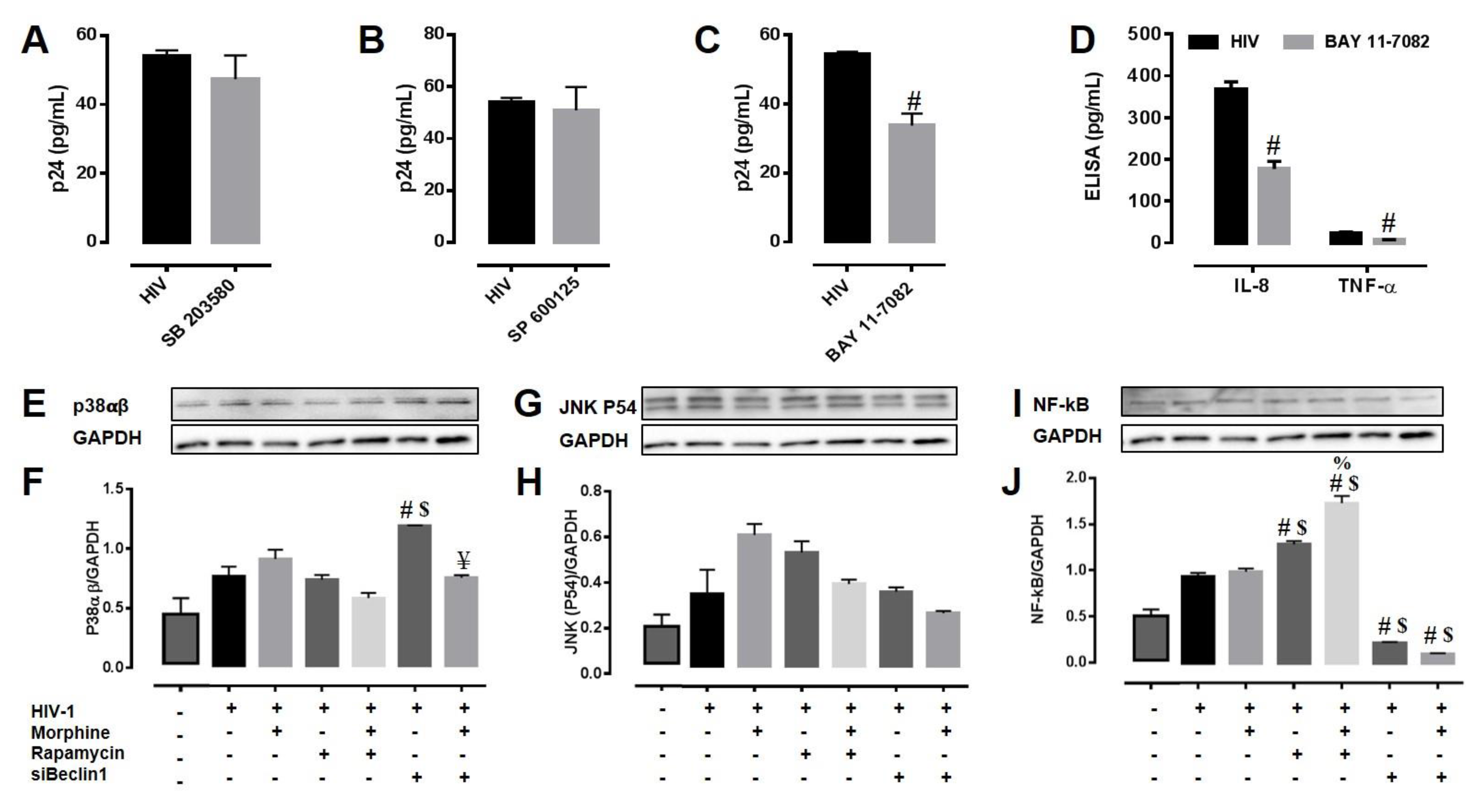
3.4. Potential Mechanism(s) Linking the Autophagy Pathway with HIV Replication and Viral-Induced Inflammation in Astrocytes
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflict of Interest
References
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 3, 1–14. [Google Scholar] [CrossRef]
- Zhou, B.Y.; Liu, Y.; Kim, B.; Xiao, Y.; He, J.J. Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol. Cell. Neurosci. 2004, 27, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Kort, J.J. Impairment of excitatory amino acid transport in astroglial cells infected with the human immunodeficiency virus type 1. AIDS Res. Hum. Retroviruses 1998, 14, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.W. Endothelial cell-astrocyte interactions. A cellular model of the blood-brain barrier. Ann. N. Y. Acad. Sci. 1988, 529, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, G.J.; Gallery, F.; MacConnell, P.; Braun, A. In situ detection of polymerase chain reaction-amplified HIV-1 nucleic acids and tumor necrosis factor-alpha RNA in the central nervous system. Am. J. Pathol. 1994, 144, 659–666. [Google Scholar] [PubMed]
- Gorry, P.R.; Ong, C.; Thorpe, J.; Bannwarth, S.; Thompson, K.A.; Gatignol, A.; Vesselingh, S.L.; Purcell, D.F. Astrocyte infection by HIV-1: Mechanisms of restricted virus replication, and role in the pathogenesis of HIV-1-associated dementia. Curr. HIV Res. 2003, 1, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; King, J.E.; Nath, A.; Calderon, T.M.; Zukin, R.S.; Bennett, M.V.; Berman, J.W. HIV-tat induces formation of an LRP-PSD-95-NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 3438–3443. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Wesselingh, S.L.; Cowley, D.; Pardo, C.A.; McArthur, J.C.; Brew, B.J.; Gorry, P.R. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann. Neurol. 2009, 66, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Conant, K.; Chen, P.; Scott, C.; Major, E.O. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J. Biol. Chem. 1999, 274, 17098–17102. [Google Scholar] [CrossRef] [PubMed]
- Gray, F.; Adle-Biassette, H.; Brion, F.; Ereau, T.; le Maner, I.; Levy, V.; Corcket, G. Neuronal apoptosis in human immunodeficiency virus infection. J. Neurovirol. 2000, 6 (Suppl. S1), S38–S43. [Google Scholar] [PubMed]
- Kaul, M.; Garden, G.A.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001, 410, 988–994. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Gurwell, J.A.; Singh, I.N.; Knapp, P.E.; Nath, A.; Hauser, K.F. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia 2005, 50, 91–106. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Wu, G.; Wang, J.; Ambati, J.; Knapp, P.E.; Reed, J.L.; Bruce-Keller, A.J.; Hauser, K.F. HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia 2006, 53, 132–146. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Bruce-Keller, A.J.; Yakovleva, T.; Bazov, I.; Bakalkin, G.; Knapp, P.E.; Hauser, K.F. Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca(2+)](i), NF-kappaB trafficking and transcription. PLoS ONE 2008, 3, e4093. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Fitting, S.; Hahn, Y.K.; Welch, S.P.; El-Hage, N.; Hauser, K.F.; Knapp, P.E. Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at mu-opioid receptor-expressing glia. Brain 2011, 134 Pt 12, 3616–3631. [Google Scholar] [CrossRef] [PubMed]
- Hauser, K.F.; Fitting, S.; Dever, S.M.; Podhaizer, E.M.; Knapp, P.E. Opiate drug use and the pathophysiology of neuroAIDS. Curr. HIV Res. 2012, 10, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.I. The Enzymatic Deacetylation Of Heroin And Closely Related Morphine Derivatives By Blood Serum. Science 1940, 92, 244–245. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antiox. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Kragh, C.L.; Ubhi, K.; Wyss-Coray, T.; Masliah, E. Autophagy in dementias. Brain Pathol. 2012, 22, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.S.; Lee, J.H. Neurodegenerative lysosomal disorders: A continuum from development to late age. Autophagy 2008, 4, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kume, S.; Kitada, M.; Kanasaki, K.; Uzu, T.; Maegawa, H.; Koya, D. Autophagy as a therapeutic target in diabetic nephropathy. Exp. Diabetes Res. 2012, 2012, 628978. [Google Scholar] [CrossRef] [PubMed]
- Nemchenko, A.; Chiong, M.; Turer, A.; Lavandero, S.; Hill, J.A. Autophagy as a therapeutic target in cardiovascular disease. J. Mol. Cell. Cardiol. 2011, 51, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.; Rubinsztein, D.C. Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 2012, 8, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, C.; Pilli, M.; Kehrl, J.H. Roles of autophagy in HIV infection. Immunol. Cell Biol. 2015, 93, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 2009, 4, e5787. [Google Scholar] [CrossRef] [PubMed]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.L.; Liao, K.; Periyasamy, P.; Yang, L.; Cai, Y.; Callen, S.E.; Buch, S. Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 2015, 11, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Rodriguez, M.; Dever, S.M.; Masvekar, R.R.; Gewirtz, D.A.; Shacka, J.J. HIV-1 and morphine regulation of autophagy in microglia: Limited interactions in the context of HIV-1 infection and opioid abuse. J. Virol. 2015, 89, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Kaushik, A.; Lapierre, J.; Dever, S.M.; El-Hage, N.; Nair, M. Electro-Magnetic Nano-Particle Bound Beclin1 siRNA Crosses the Blood-Brain Barrier to Attenuate the Inflammatory Effects of HIV-1 Infection in Vitro. J. Neuroimmune Pharmacol. 2016, 12, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Hormonally active vitamin D3 (1alpha,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J. Biol. Chem. 2011, 286, 18890–18902. [Google Scholar] [CrossRef] [PubMed]
- Pomilio, C.; Pavia, P.; Gorojod, R.M.; Vinuesa, A.; Alaimo, A.; Galvan, V.; Kotler, M.L.; Beauquis, J.; Saravia, F. Glial alterations from early to late stages in a model of Alzheimer’s disease: Evidence of autophagy involvement in Aβ internalization. Hippocampus 2016, 26, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Fryer, J.D.; Settembre, C.; Ballabio, A. Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorder. Proc. Natl. Acad. Sci. USA 2012, 109, E2334–E2342. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrian, C.; Mosharov, E.V.; Tang, G.; Cheng, H.C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012, 74, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Weston, M.C.; Chen, H.; Swann, J.W. Multiple roles for mammalian target of rapamycin signaling in both glutamatergic and GABAergic synaptic transmission. J. Neurosci. 2012, 32, 11441–11452. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lu, T.; Yue, X.; Wei, N.; Jiang, Y.; Chen, M.; Ni, G.; Liu, X.; Xu, G. Neuroprotective effect of ginsenoside Rb1 on glutamate-induced neurotoxicity: With emphasis on autophagy. Neurosci. Lett. 2010, 482, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Bigford, G.E.; Alonso, O.F.; Dietrich, D.; Keane, R.W. A novel protein complex in membrane rafts linking the NR2B glutamate receptor and autophagy is disrupted following traumatic brain injury. J. Neurotrauma 2009, 26, 703–720. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.J.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Smaili, S.S. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef] [PubMed]
- Stamoula, E.; Vavilis, T.; Aggelidou, E.; Kaidoglou, A.; Cheva, A.; Mellidis, K.; Lazou, A.; Haitoglou, C.; Albani, M.; Kritis, A. Low Dose Administration of Glutamate Triggers a Non-Apoptotic, Autophagic Response in PC12 Cells. Cell. Phys. Biochem. 2015, 37, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Mehla, R.; Chauhan, A. HIV-1 differentially modulates autophagy in neurons and astrocytes. J. Neuroimmunol. 2015, 285, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Fu, M.; Kumar, S.; Kumar, A. Methamphetamine potentiates HIV-1 gp120-mediated autophagy via Beclin-1 and Atg5/7 as a pro-survival response in astrocytes. Cell Death Dis. 2016, 7, e2425. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Dever, S.M.; Fitting, S.; Ahmed, T.; Hauser, K.F. HIV-1 coinfection and morphine coexposure severely dysregulate hepatitis C virus-induced hepatic proinflammatory cytokine release and free radical production: Increased pathogenesis coincides with uncoordinated host defenses. J. Virol. 2011, 85, 11601–11614. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Dever, S.M.; Podhaizer, E.M.; Arnatt, C.K.; Zhang, Y.; Hauser, K.F. A novel bivalent HIV-1 entry inhibitor reveals fundamental differences in CCR5-μ-opioid receptor interactions between human astroglia and microglia. AIDS 2013, 27, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Rodriguez, M.; Podhaizer, E.M.; Zou, S.; Dever, S.M.; Snider, S.E.; Knapp, P.E.; Beardsley, P.M.; Hauser, K.F. Ibudilast (AV411), and its AV1013 analog, reduce HIV-1 replication and neuronal death induced by HIV-1 and morphine. AIDS 2014, 28, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Fitting, S.; Zou, S.; El-Hage, N.; Suzuki, M.; Paris, J.J.; Schier, C.J.; Rodriguez, J.W.; Rodriguez, M.; Knapp, P.E.; Hauser, K.F. Opiate addiction therapies and HIV-1 Tat: Interactive effects on glial [Ca(2+)]i, oxyradical and neuroinflammatory chemokine production and correlative neurotoxicity. Curr. HIV Res. 2014, 12, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Holden, C.P.; Nath, A.; Geiger, J.D. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein Tat. J. Neurochem. 1999, 73, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Padua, R.A.; Geiger, J.D. HIV-1 coat protein gp120-induced increases in levels of intrasynaptosomal calcium. Brain Res. 1995, 678, 200–206. [Google Scholar] [CrossRef]
- Haughey, N.J.; Mattson, M.P. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J. Acquir. Immune Defic. Syndr. 2002, 31 (Suppl. S2), S55–S61. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Calcium channel antagonists and human immunodeficiency virus coat protein-mediated neuronal injury. Ann. Neurol. 1991, 30, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Holden, C.P.; Haughey, N.J.; Nath, A.; Geiger, J.D. Role of Na+/H+ exchangers, excitatory amino acid receptors and voltage-operated Ca2+ channels in human immunodeficiency virus type 1 gp120-mediated increases in intracellular Ca2+ in human neurons and astrocytes. Neuroscience 1999, 91, 1369–1378. [Google Scholar] [CrossRef]
- Bai, X.; Jiang, Y. Key factors in mTOR regulation. Cell. Mol. Life Sci. 2010, 67, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, J.R.; Mulcahy Levy, J.M.; Coultrap, S.J.; Thorburn, A.; Bayer, K.U. Excitotoxic glutamate insults block autophagic flux in hippocampal neurons. Brain Res. 2014, 1542, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Osiecki, K.; Lopez, L.; Goldstein, H.; Calderon, T.M.; Berman, J.W. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: A potential mechanism of HIV-CNS invasion and NeuroAIDS. J. Neurosci. 2006, 26, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Airoldi, M.; Bandera, A.; Trabattoni, D.; Tagliabue, B.; Arosio, B.; Soria, A.; Rainone, V.; Lapadula, G.; Annoni, G.; Clerici, M.; et al. Neurocognitive impairment in HIV-infected naive patients with advanced disease: The role of virus and intrathecal immune activation. Clin. Dev. Immunol. 2012, 2012, 467154. [Google Scholar] [CrossRef] [PubMed]
- Fiume, G.; Vecchio, E.; De Laurentiis, A.; Trimboli, F.; Palmieri, C.; Pisano, A.; Falcone, C.; Pontoriero, M.; Rossi, A.; Scialdone, A.; et al. Human immunodeficiency virus-1 Tat activates NF-κB via physical interaction with IκB-α and p65. Nucleic Acids Res. 2012, 40, 3548–3562. [Google Scholar] [CrossRef] [PubMed]
- Pitha, P.M. Innate antiviral response: Role in HIV-1 infection. Viruses 2011, 3, 1179–1203. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.P.; Su, Y.C.; Lee, P.H.; Lei, H.Y. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy 2013, 9, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Cornell-Bell, A.H.; Finkbeiner, S.M.; Cooper, M.S.; Smith, S.J. Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science 1990, 247, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.C.; Merrill, J.E.; Dirksen, E.R.; Sanderson, M.J. Intercellular signaling in glial cells: Calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 1991, 6, 983–992. [Google Scholar] [CrossRef]
- Dani, J.W.; Chernjavsky, A.; Smith, S.J. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron 1992, 8, 429–440. [Google Scholar] [CrossRef]
- Porter, J.T.; McCarthy, K.D. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J. Neurosci. 1996, 16, 5073–5081. [Google Scholar] [PubMed]
- Lipton, S.A. HIV-related neuronal injury. Potential therapeutic intervention with calcium channel antagonists and NMDA antagonists. Mol. Neurobiol. 1994, 8, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Haughey, N.J.; Jones, M.; Anderson, C.; Bell, J.E.; Geiger, J.D. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: Protection by memantine. Ann. Neurol. 2000, 47, 186–194. [Google Scholar] [CrossRef]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte-neuron signalling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Sanzgiri, R.P.; Parpura, V.; Haydon, P.G. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J. Neurosci. 1998, 18, 6822–6829. [Google Scholar] [PubMed]
- Bezzi, P.; Carmignoto, G.; Pasti, L.; Vesce, S.; Rossi, D.; Rizzini, B.L.; Pozzan, T.; Volterra, A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 1998, 391, 281–285. [Google Scholar] [PubMed]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pekarskaya, O.; Bencheikh, M.; Chao, W.; Gelbard, H.A.; Ghorpade, A.; Rothstein, J.D.; Volsky, D.J. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology 2003, 312, 60–73. [Google Scholar] [CrossRef]
- Chen, L.L.; Wu, J.C.; Wang, L.H.; Wang, J.; Qin, Z.H.; Difiglia, M.; Lin, F. Rapamycin prevents the mutant huntingtin-suppressed GLT-1 expression in cultured astrocytes. Acta Pharmacol. Sin. 2012, 33, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heredia, A.; Amoroso, A.; Davis, C.; Le, N.; Reardon, E.; Dominique, J.K.; Klingebiel, E.; Gallo, R.C.; Redfield, R.R. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV β-chemokines: An approach to suppress R5 strains of HIV-1. Proc. Natl. Acad. Sci. USA 2003, 100, 10411–10416. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.D.; Taub, D.D.; Perry, V.H. Overriding the brain’s intrinsic resistance to leukocyte recruitment with intraparenchymal injections of recombinant chemokines. Neuroscience 1996, 74, 283–292. [Google Scholar] [CrossRef]
- Gouwy, M.; Struyf, S.; Noppen, S.; Schutyser, E.; Springael, J.Y.; Parmentier, M.; Proost, P.; Van Damme, J. Synergy between coproduced CC and CXC chemokines in monocyte chemotaxis through receptor-mediated events. Mol. Pharmacol. 2008, 74, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Gerszten, R.E.; Garcia-Zepeda, E.A.; Lim, Y.C.; Yoshida, M.; Ding, H.A.; Gimbrone, M.A., Jr.; Luster, A.D.; Luscinskas, F.W.; Rosenzweig, A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 1999, 398, 718–723. [Google Scholar] [PubMed]
- Ott, M.; Lovett, J.L.; Mueller, L.; Verdin, E. Superinduction of IL-8 in T cells by HIV-1 Tat protein is mediated through NF-κB factors. J. Immunol. 1998, 160, 2872–2880. [Google Scholar] [PubMed]
- Kutsch, O.; Oh, J.; Nath, A.; Benveniste, E.N. Induction of the chemokines interleukin-8 and IP-10 by human immunodeficiency virus type 1 Tat in astrocytes. J. Virol. 2000, 74, 9214–9221. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Kumar, A. HIV-1 gp120-mediated increases in IL-8 production in astrocytes are mediated through the NF-κB pathway and can be silenced by gp120-specific siRNA. J. Neuroinflamm. 2010, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Youn, G.S.; Kwon, D.J.; Ju, S.M.; Rhim, H.; Bae, Y.S.; Choi, S.Y.; Park, J. Celastrol ameliorates HIV-1 Tat-induced inflammatory responses via NF-κB and AP-1 inhibition and heme oxygenase-1 induction in astrocytes. Toxicol. Appl. Pharmacol. 2014, 280, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.C.; Huang, Y.; Tang, K.; Cui, M.; Niemann, D.; Lopez, A.; Morgello, S.; Chen, S. HIV-1-infected and/or immune-activated macrophages regulate astrocyte CXCL8 production through IL-1β and TNF-α: Involvement of mitogen-activated protein kinases and protein kinase R. J. Neuroimmunol. 2008, 200, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Gangwani, M.R.; Kumar, A. Multiple Protein Kinases via Activation of Transcription Factors NF-κB, AP-1 and C/EBP-δ Regulate the IL-6/IL-8 Production by HIV-1 Vpr in Astrocytes. PLoS ONE 2015, 10, e0135633. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L.; Baeuerle, P.A. Endoplasmicreticulum-induced signal transduction and gene expression. Trends Cell Biol. 1997, 7, 50–55. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Inflammation in Alzheimer’s disease: Amyloid-β oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 2009, 87, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Yan, P.; Qu, Z.; Liu, H.; Xiao, G. Hsp90 regulates processing of NF-kappa B2 p100 involving protection of NF-kappa B-inducing kinase (NIK) from autophagy-mediated degradation. Cell Res. 2007, 17, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. Doctor Jekyll and Mister Hyde: Autophagy can promote both cell survival and cell death. Cell Death Differ. 2005, 12 (Suppl. S2), 1468–1472. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, M.; Lapierre, J.; Ojha, C.R.; Estrada-Bueno, H.; Dever, S.M.; Gewirtz, D.A.; Kashanchi, F.; El-Hage, N. Importance of Autophagy in Mediating Human Immunodeficiency Virus (HIV) and Morphine-Induced Metabolic Dysfunction and Inflammation in Human Astrocytes. Viruses 2017, 9, 201. https://doi.org/10.3390/v9080201
Rodriguez M, Lapierre J, Ojha CR, Estrada-Bueno H, Dever SM, Gewirtz DA, Kashanchi F, El-Hage N. Importance of Autophagy in Mediating Human Immunodeficiency Virus (HIV) and Morphine-Induced Metabolic Dysfunction and Inflammation in Human Astrocytes. Viruses. 2017; 9(8):201. https://doi.org/10.3390/v9080201
Chicago/Turabian StyleRodriguez, Myosotys, Jessica Lapierre, Chet Raj Ojha, Hary Estrada-Bueno, Seth M. Dever, David A. Gewirtz, Fatah Kashanchi, and Nazira El-Hage. 2017. "Importance of Autophagy in Mediating Human Immunodeficiency Virus (HIV) and Morphine-Induced Metabolic Dysfunction and Inflammation in Human Astrocytes" Viruses 9, no. 8: 201. https://doi.org/10.3390/v9080201
APA StyleRodriguez, M., Lapierre, J., Ojha, C. R., Estrada-Bueno, H., Dever, S. M., Gewirtz, D. A., Kashanchi, F., & El-Hage, N. (2017). Importance of Autophagy in Mediating Human Immunodeficiency Virus (HIV) and Morphine-Induced Metabolic Dysfunction and Inflammation in Human Astrocytes. Viruses, 9(8), 201. https://doi.org/10.3390/v9080201