
Characterization of Two Historic Smallpox Specimens from a Czech Museum

 ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Diagnostic Specimens
2.2. Microscopy
2.3. DNA Extraction and PCR Analysis
2.4. Age Estimation of Specimens and Composition of the Fixative Solution
2.5. Next Generation Sequencing Analysis and Phylogenetic Analysis
2.6. Estimation of Molecular Clock
2.7. High-Resolution Mass Spectrometry
3. Results
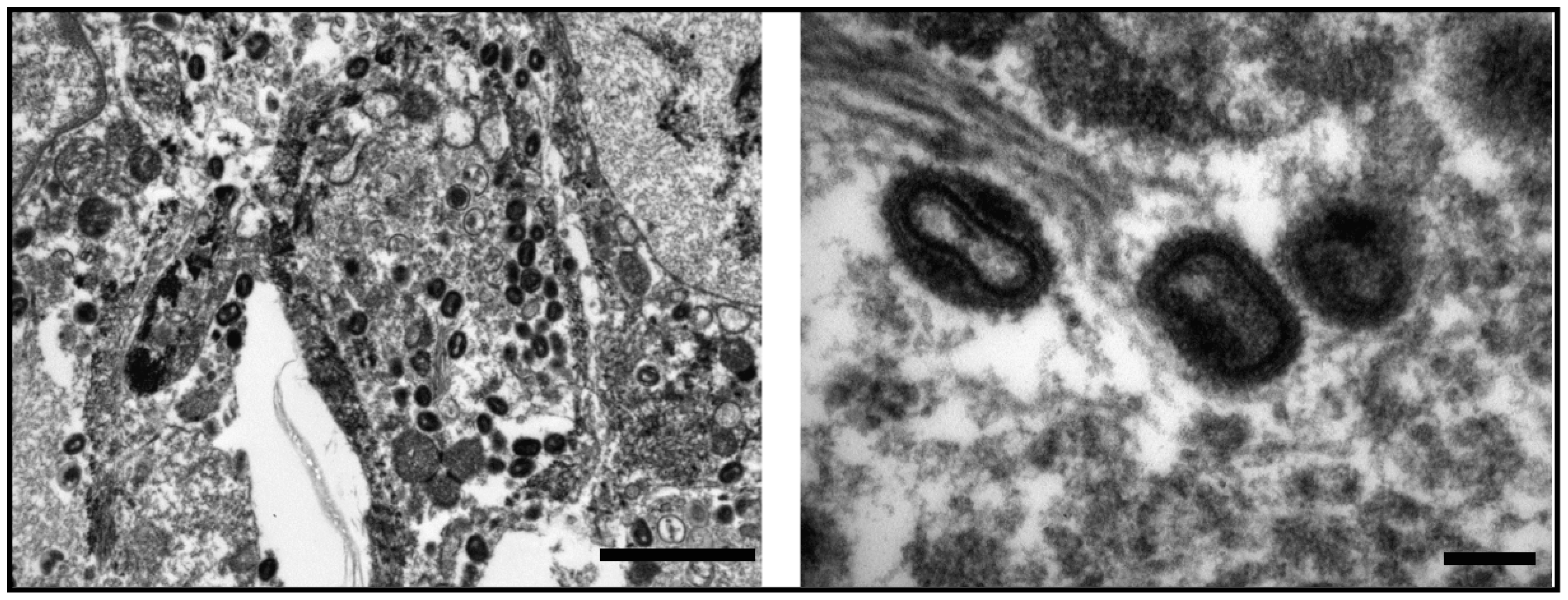
3.1. Diagnostic Specimens and Microscopy
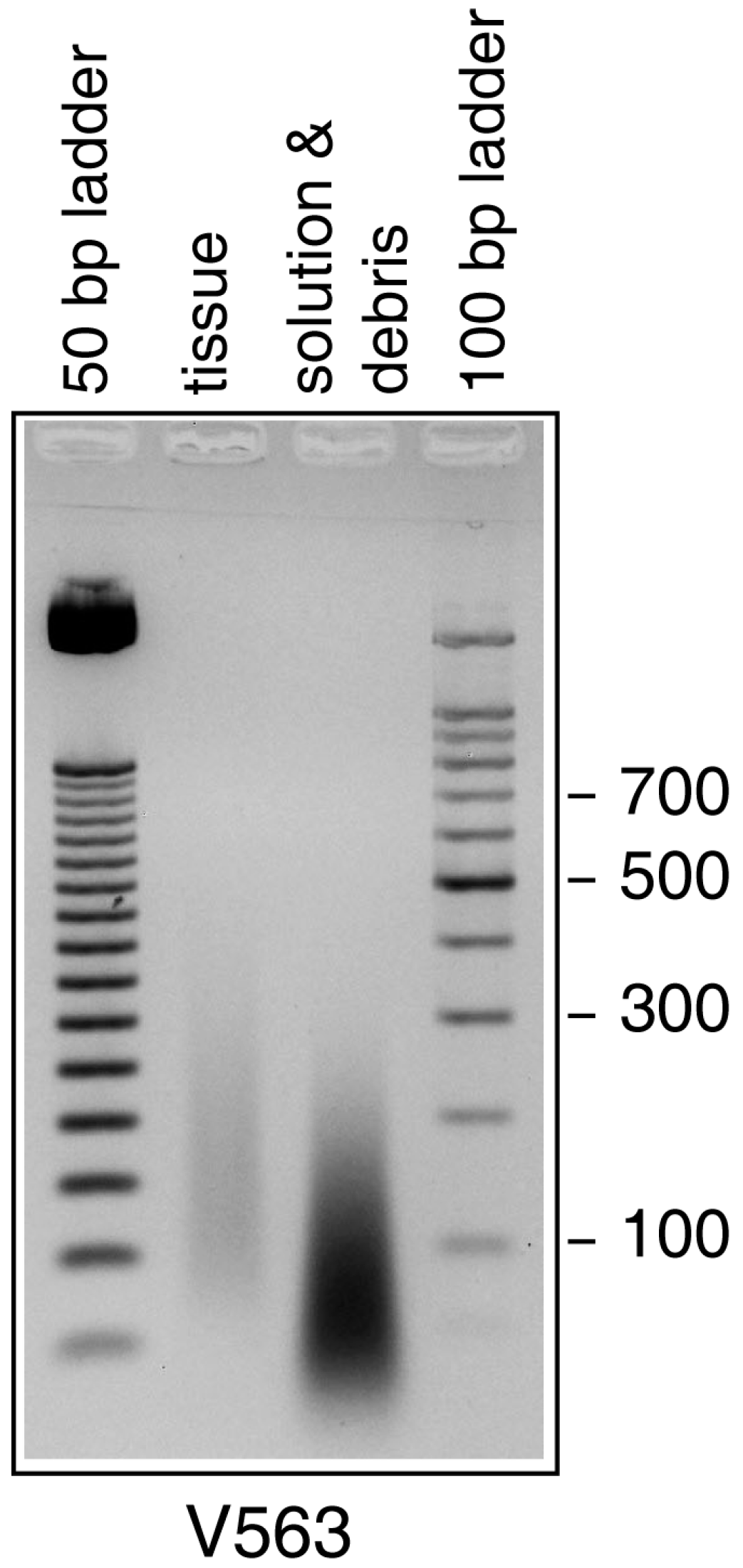
3.2. Detection of Variola virus DNA
3.3. Age of Specimens and Composition of the Fixative Solution
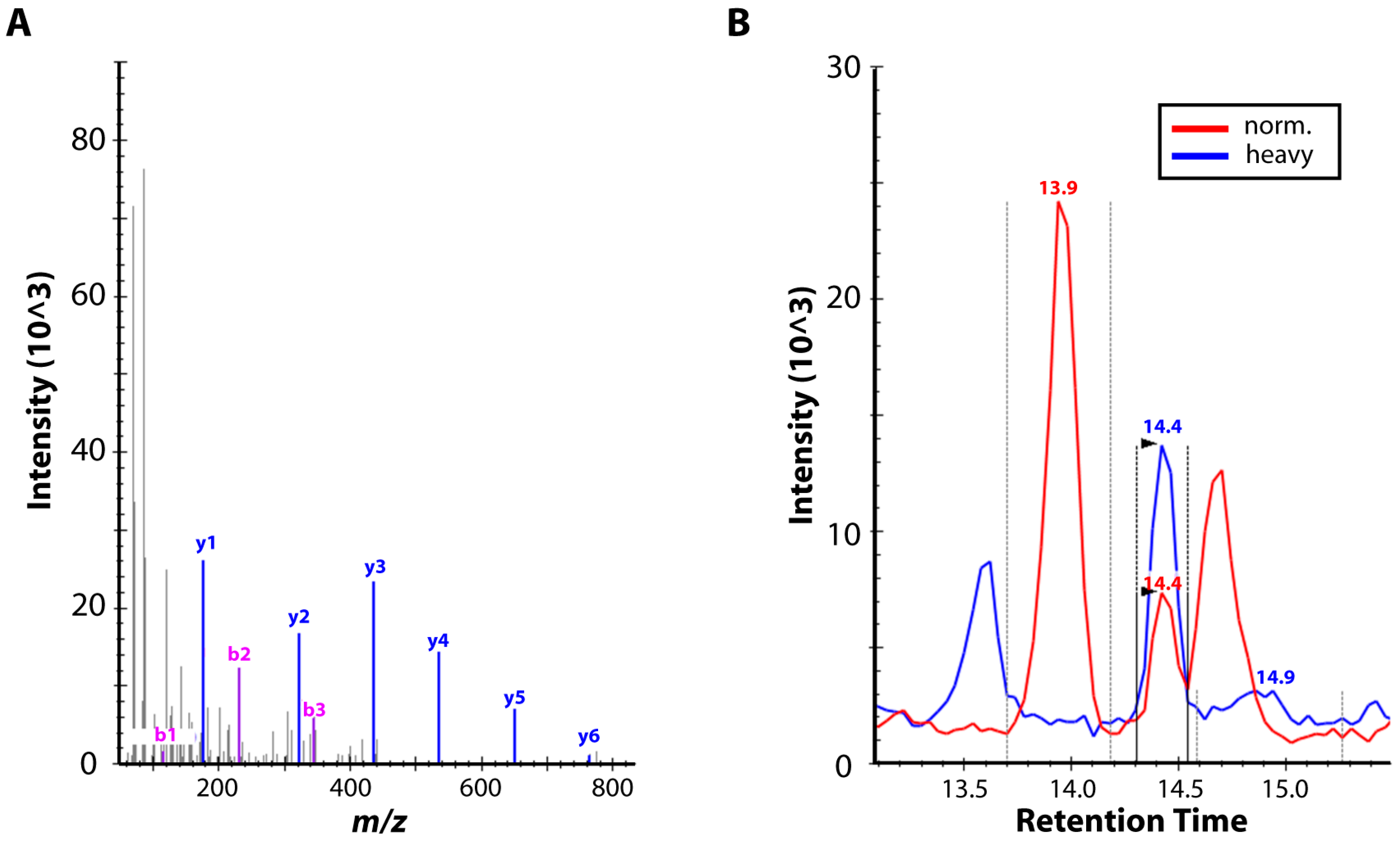
3.4. High-Resolution Mass Spectrometry
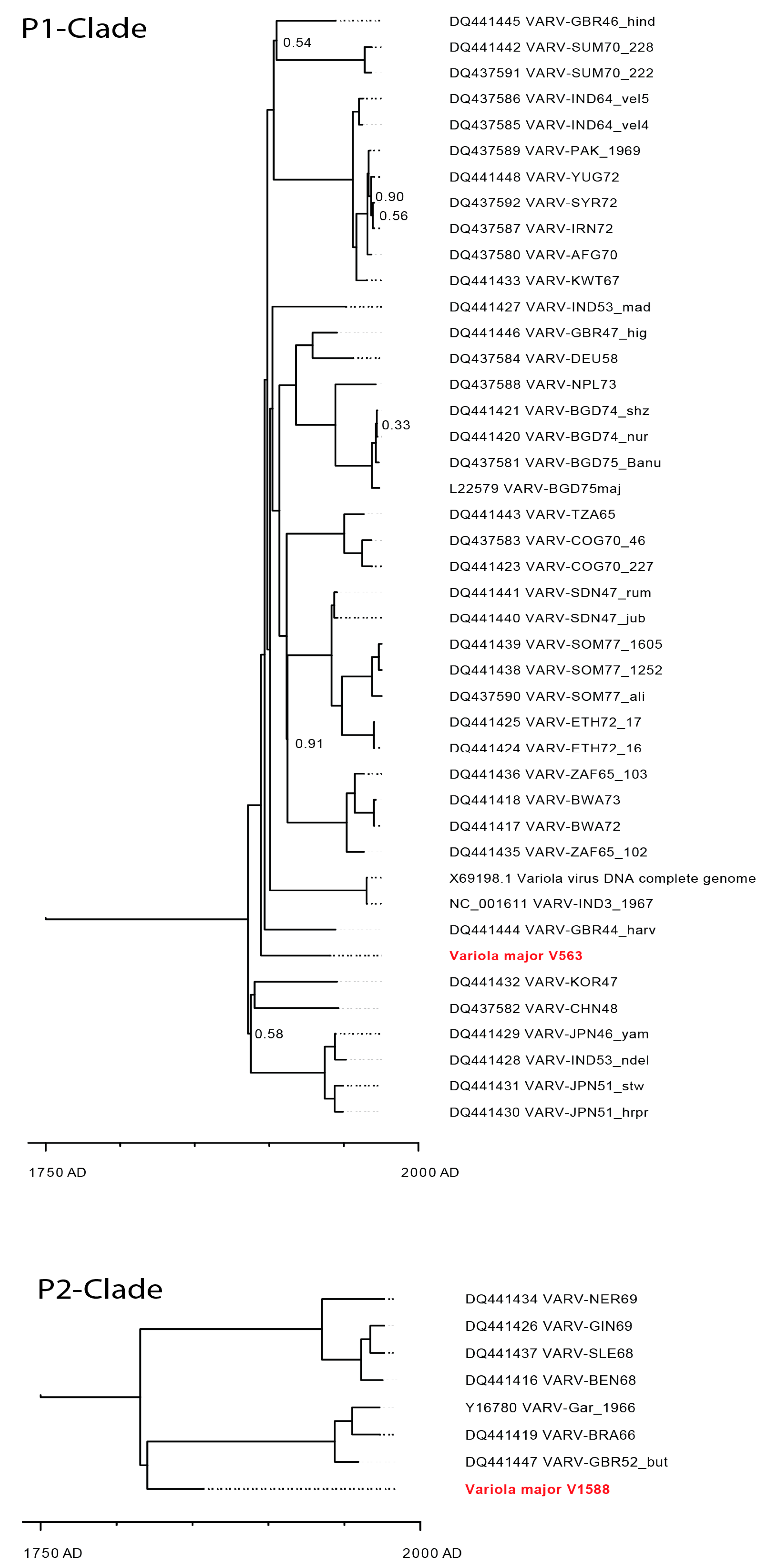
3.5. Next Generation Sequencing and Phylogenetic Analysis
3.6. Molecular Clock Comparison
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. Analysis of the Preservative Solutions
References
- Fenner, F.; Henderson, D.A.; Arita, I.; Jezek, Z.; Ladnyi, I.D. Smallpox; World Health Organ: Geneva, Switzerland, 1988. [Google Scholar]
- Geddes, A.M. The history of smallpox. Clin. Dermatol. 2006, 24, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.A.; Arita, I. The Smallpox Threat: A Time to Reconsider Global Policy. Biosecur. Bioterror. Biodef. Strateg. Pract. Sci. 2014, 12, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Esposito, J.J. Genome Sequence Diversity and Clues to the Evolution of Variola (Smallpox) Virus. Science 2006, 313, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Babkin, V.I.; Babkina, N.I. A retrospective study of the orthopoxvirus molecular evolution. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2012, 12, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Babkin, I.V.; Nepomnyashchikh, T.S.; Maksyutov, R.A.; Gutorov, V.V.; Babkina, I.N.; Shchelkunov, S.N. Comparative analysis of variable regions in the variola virus genome. Mol. Biol. 2008, 42, 543–553. [Google Scholar] [CrossRef]
- Li, Y.; Carroll, D.S.; Gardner, S.N.; Walsh, M.C.; Vitalis, E.A.; Damon, I.K. On the origin of smallpox: Correlating variola phylogenics with historical smallpox records. Proc. Natl. Acad. Sci. USA 2007, 104, 15787–15792. [Google Scholar] [CrossRef] [PubMed]
- Babkin, V.I.; Shchelkunov, N.S. [Molecular evolution of poxviruses]. Genetika 2008, 44, 1029–1044. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov, S.N. How long ago did smallpox virus emerge? Arch. Virol. 2009, 154, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Duggan, A.T.; Perdomo, M.F.; Piombino-Mascali, D.; Marciniak, S.; Poinar, D.; Emery, M.V.; Buchmann, J.P.; Duchêne, S.; Jankauskas, R.; Humphreys, M.; et al. 17th Century Variola Virus Reveals the Recent History of Smallpox. Curr. Biol. 2016, 26, 3407–3412. [Google Scholar] [CrossRef] [PubMed]
- Biagini, P.; Thèves, C.; Balaresque, P.; Géraut, A.; Cannet, C.; Keyser, C.; Nikolaeva, D.; Gérard, P.; Duchesne, S.; Orlando, L.; et al. Variola Virus in a 300-Year-Old Siberian Mummy. N. Engl. J. Med. 2012, 367, 2057–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enserink, M.; Stone, R. Dead Virus Walking. Science 2002, 295, 2001–2005. [Google Scholar] [CrossRef] [PubMed]
- Fornaciari, G.; Marchetti, A. Intact smallpox virus particles in an Italian mummy of sixteenth century. Lancet 1986, 328, 625. [Google Scholar] [CrossRef]
- McCollum, A.M.; Li, Y.; Wilkins, K.; Karem, K.L.; Davidson, W.B.; Paddock, C.D.; Reynolds, M.G.; Damon, I.K. Poxvirus Viability and Signatures in Historical Relics. Emerg. Infect. Dis. 2014, 20, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Schoepp, R.J.; Morin, M.D.; Martinez, M.J.; Kulesh, D.A.; Hensley, L.; Geisbert, T.W.; Brady, D.R.; Jahrling, P.B. Detection and identification of Variola virus in fixed human tissue after prolonged archival storage. Lab. Investig. 2004, 84, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Thèves, C.; Biagini, P.; Crubézy, E. The rediscovery of smallpox. Clin. Microbiol. Infect. 2014, 20, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Smrčka, V.; Kuželka, V.; Povýšil, C. Introduction. In Atlas of Diseases in Dry Bones: Upper and Lower Extremities; Academia: Prague, Czech Republic, 2009; pp. 15–33. [Google Scholar]
- Fedele, C.G.; Negredo, A.; Molero, F.; Sanchez-Seco, M.P.; Tenorio, A. Use of Internally Controlled Real-Time Genome Amplification for Detection of Variola Virus and Other Orthopoxviruses Infecting Humans. J. Clin. Microbiol. 2006, 44, 4464–4470. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.V.; Laue, T.; Laker, T.M.; Babkin, V.I.; Drosten, C.; Shchelkunov, N.S.; Niedrig, M.; Damon, K.I.; Meyer, H. Real-time PCR system for detection of orthopoxviruses and simultaneous identification of smallpox virus. J. Clin. Microbiol. 2004, 42, 1940–1946. [Google Scholar] [CrossRef] [PubMed]
- Damaso, C.R.A.; Esposito, J.J.; Condit, R.C.; Moussatché, N. An Emergent Poxvirus from Humans and Cattle in Rio de Janeiro State: Cantagalo Virus May Derive from Brazilian Smallpox Vaccine. Virology 2000, 277, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Pilin, A.; Čabala, R.; Pudil, F.; Bencko, V. The Use of the d-, l- Aspartic Ratio in Decalcified Collagen from Human Dentin as an Estimator of Human Age. J. Forensic Sci. 2001, 46, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM; ArXiv13033997 Q-Bio; Cornell University Library: Ithaca, NY, USA, 2013. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, S.A.; Lesin, M.V.; Nikolenko, I.S.; Pham, S.; Prjibelski, D.A.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.B.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, A.C.; Zeng, Q.; Wortman, J.; Young, K.S.; Earl, M.A. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Upton, C.; Slack, S.; Hunter, A.L.; Ehlers, A.; Roper, R.L. Poxvirus Orthologous Clusters: toward Defining the Minimum Essential Poxvirus Genome. J. Virol. 2003, 77, 7590–7600. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Mann, M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 2009, 8, 5674–5678. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinform. Oxf. Engl. 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Massung, R.F.; Knight, J.C.; Esposito, J.J. Topography of Variola Smallpox Virus Inverted Terminal Repeats. Virology 1995, 211, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Wang, C.; Upton, C. Poxviruses: Past, present and future. Virus Res. 2006, 117, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Smithson, C.; Purdy, A.; Verster, A.J.; Upton, C. Prediction of Steps in the Evolution of Variola Virus Host Range. PLoS ONE 2014, 9, e91520. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Yang, W.; McKinney, S.; Kalloger, S.; Gale, N.; Ng, Y.; Chow, K.; Bell, L.; Lorette, J.; Carrier, M.; et al. Nucleic acid quantity and quality from paraffin blocks: defining optimal fixation, processing and DNA/RNA extraction techniques. Exp. Mol. Pathol. 2012, 92, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Howat, W.J.; Wilson, B.A. Tissue fixation and the effect of molecular fixatives on downstream staining procedures. Methods (San Diego Calif.) 2014, 70, 12–19. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Protein ID | Peptide | Specificity |
|---|---|---|---|
| VLFT-4 viral late transcription factor OS | Q0N822_VARV | ISAVSTVLEDVQAAGISR | Orthopoxvirus |
| ORF1L (Fragment) | Q89160_VARV | YQSLIPRLGINYLIDTTSR | Orthopoxvirus |
| Core protein VP8 | VP8_VAR67 | SmLSIFNIVPR | Orthopoxvirus |
| 14 kDa protein | K7ZBW7_VARV | NDDVLFR | Variola virus |
| Cowpox A-type inclusion protein | Q0NBD3_VARV | VLLLTPEVASLR | Orthopoxvirus |
| Late transcription factor 1 | VLTF1_VAR67 | VNVFETR | Orthopoxvirus |
| Cowpox A-type inclusion protein | Q0NBD3_VARV | ISDLER | Orthopoxvirus |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pajer, P.; Dresler, J.; Kabíckova, H.; Písa, L.; Aganov, P.; Fucik, K.; Elleder, D.; Hron, T.; Kuzelka, V.; Velemínsky, P.; et al. Characterization of Two Historic Smallpox Specimens from a Czech Museum. Viruses 2017, 9, 200. https://doi.org/10.3390/v9080200
Pajer P, Dresler J, Kabíckova H, Písa L, Aganov P, Fucik K, Elleder D, Hron T, Kuzelka V, Velemínsky P, et al. Characterization of Two Historic Smallpox Specimens from a Czech Museum. Viruses. 2017; 9(8):200. https://doi.org/10.3390/v9080200
Chicago/Turabian StylePajer, Petr, Jiri Dresler, Hana Kabíckova, Libor Písa, Pavel Aganov, Karel Fucik, Daniel Elleder, Tomas Hron, Vitezslav Kuzelka, Petr Velemínsky, and et al. 2017. "Characterization of Two Historic Smallpox Specimens from a Czech Museum" Viruses 9, no. 8: 200. https://doi.org/10.3390/v9080200
APA StylePajer, P., Dresler, J., Kabíckova, H., Písa, L., Aganov, P., Fucik, K., Elleder, D., Hron, T., Kuzelka, V., Velemínsky, P., Klimentova, J., Fucikova, A., Pejchal, J., Hrabakova, R., Benes, V., Rausch, T., Dundr, P., Pilin, A., Cabala, R., ... Meyer, H. (2017). Characterization of Two Historic Smallpox Specimens from a Czech Museum. Viruses, 9(8), 200. https://doi.org/10.3390/v9080200