
Evaluation of Taterapox Virus in Small Animals


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cells and Viruses
2.3. Psoralen Inactivation
2.4. ELISAs
2.5. In-Life Animal Assays, Cytokine Analysis and qPCR
2.6. Microscopy
2.7. Histopathology
2.8. Statistics
3. Results
3.1. TATV Infection of Immunocompetent Animal
3.2. Inactivated TATV Induces Seroconversion
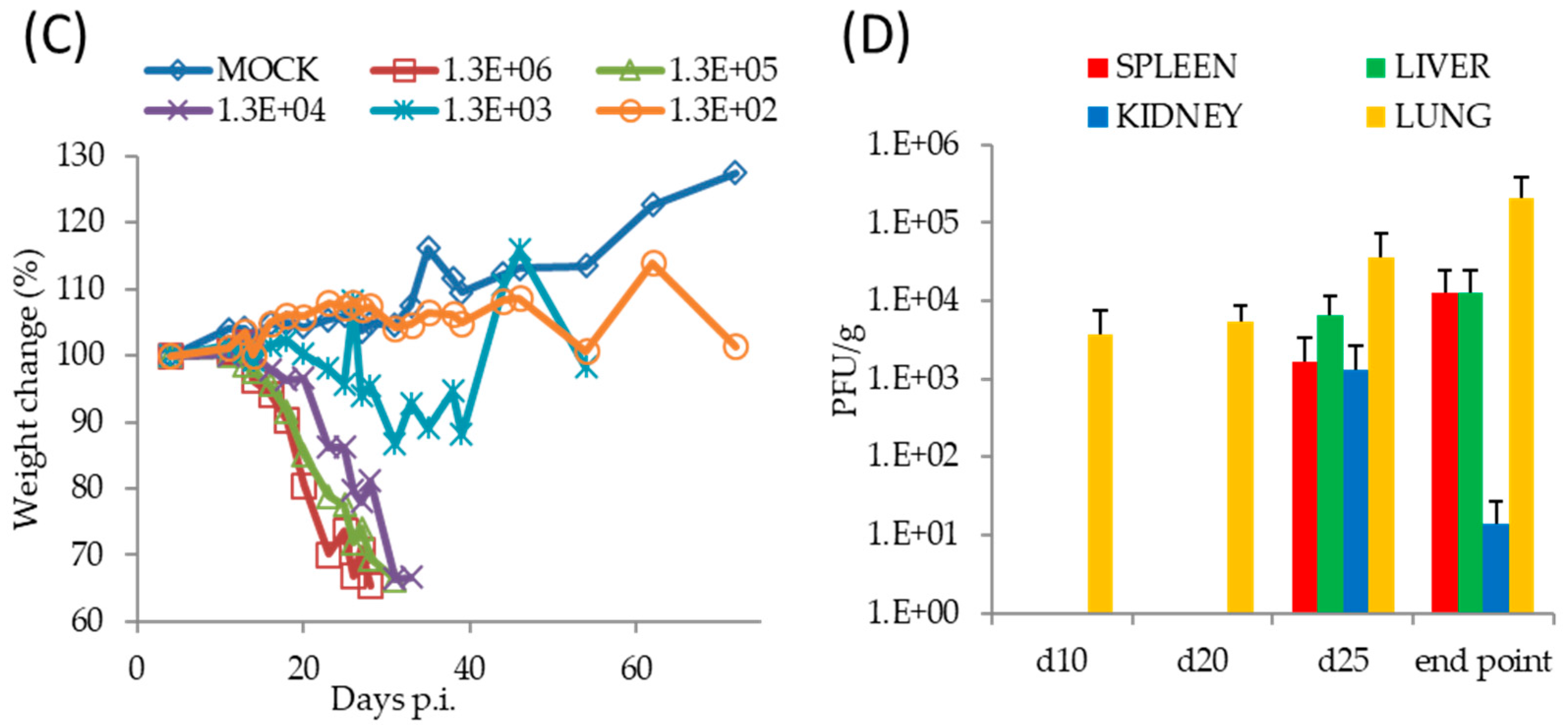
3.3. TATV Infection of Immunodeficient Mice
3.4. Mortality in SCID Mice Is Not a Function of Virus Mutation
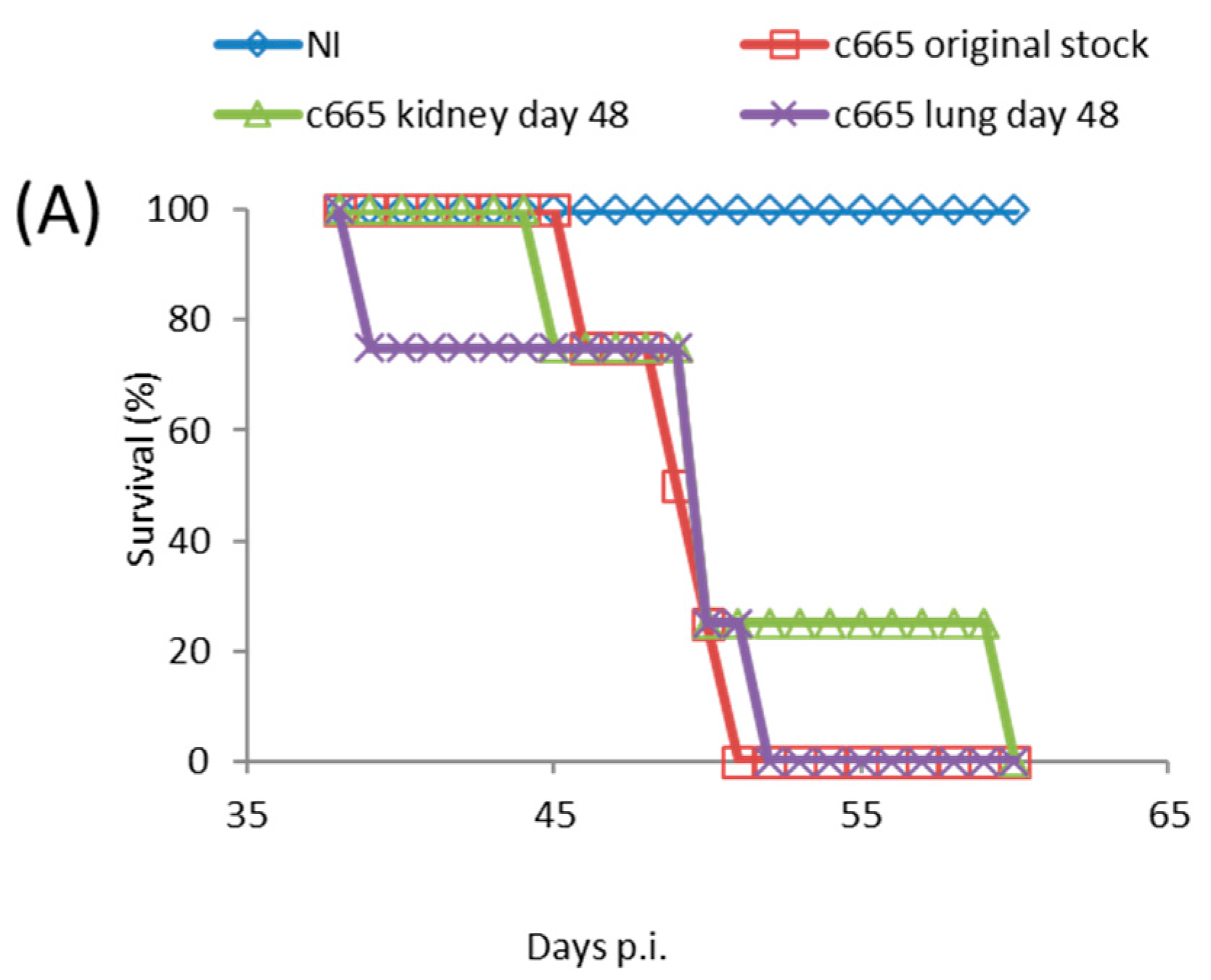
3.5. TATV Transmission
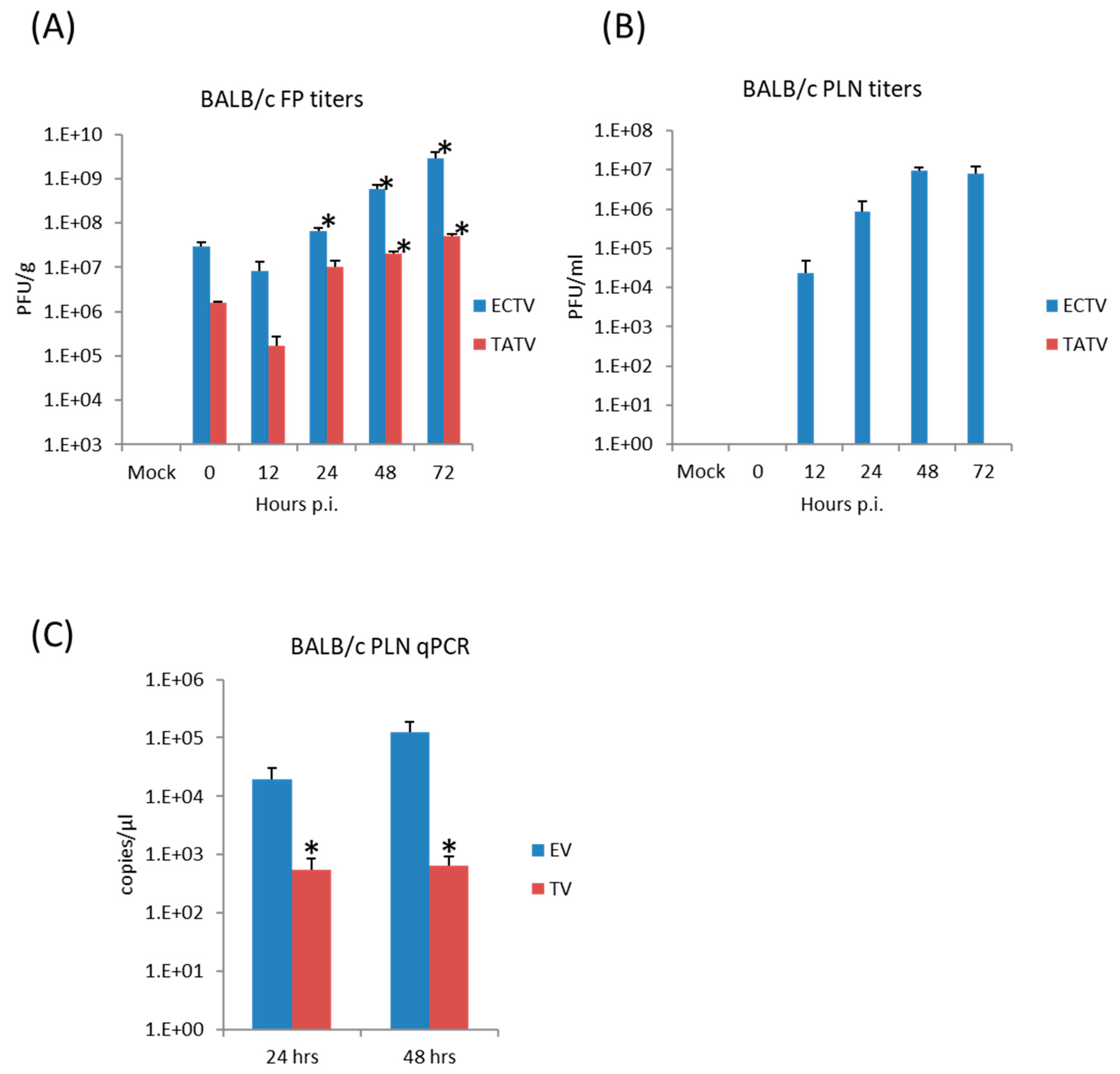
3.6. Virus Replication and Cytokine Synthesis in the Primary Site of Infection (Footpad) and the Draining (Popliteal) Lymph Node
3.7. The Host Response at the Draining Lymph Node
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fenner, F. The pathogenesis of the acute exanthems; an interpretation based on experimental investigations with mousepox; infectious ectromelia of mice. Lancet 1948, 2, 915–920. [Google Scholar] [CrossRef]
- Fenner, F. The biological characters of several strains of vaccinia, cowpox and rabbitpox viruses. Virology 1958, 5, 502–529. [Google Scholar] [CrossRef]
- Mims, C.A. The response of mice to the intravenous injection of cowpox virus. Br. J. Exp. Pathol. 1968, 49, 24–32. [Google Scholar] [PubMed]
- Williamson, J.D.; Reith, R.W.; Jeffrey, L.J.; Arrand, J.R.; Mackett, M. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J. Gen. Virol. 1990, 71 Pt 11, 2761–2767. [Google Scholar] [CrossRef]
- Bedson, H.S.; Duckworth, M.J. Rabbit pox: An experimental study of the pathways of infection in rabbits. J. Pathol. Bacteriol. 1963, 85, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, N.; Roper, R.L.; Feng, Z.; Hunter, A.; Danila, M.; Lefkowitz, E.J.; Buller, R.M.; Upton, C. Complete coding sequences of the rabbitpox virus genome. J. Gen. Virol. 2005, 86, 2969–2977. [Google Scholar] [CrossRef] [PubMed]
- Nalca, A.; Livingston, V.A.; Garza, N.L.; Zumbrun, E.E.; Frick, O.M.; Chapman, J.L.; Hartings, J.M. Experimental infection of cynomolgus macaques (Macaca fascicularis) with aerosolized monkeypox virus. PLoS ONE 2010, 5, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, R.J.; Smedley, J.V.; Perera, P.Y.; Silvera, P.M.; Waldmann, T.A.; Capala, J.; Perera, L.P. Smallpox vaccine with integrated IL-15 demonstrates enhanced in vivo viral clearance in immunodeficient mice and confers long term protection against a lethal monkeypox inoculation in cynomolgus monkeys. Vaccine 2010, 28, 7081–7091. [Google Scholar] [CrossRef] [PubMed]
- Stittelaar, K.J.; Neyts, J.; Naesens, L.; van Amerongen, G.; van Lavieren, R.F.; Holy, A.; De Clercq, E.; Niesters, H.G.; Fries, E.; Maas, C.; et al. Antiviral treatment is more effective than smallpox vaccination upon lethal monkeypox virus infection. Nature 2006, 439, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Heraud, J.M.; Edghill-Smith, Y.; Ayala, V.; Kalisz, I.; Parrino, J.; Kalyanaraman, V.S.; Manischewitz, J.; King, L.R.; Hryniewicz, A.; Trindade, C.J.; et al. Subunit recombinant vaccine protects against monkeypox. J. Immunol. 2006, 177, 2552–2564. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.W.; Thompson, E.; Wilhelmsen, C.; Zimmerman, M.; Ichou, M.A.; Steffen, S.E.; Schmaljohn, C.S.; Schmaljohn, A.L.; Jahrling, P.B. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J. Virol. 2004, 78, 4433–4443. [Google Scholar] [CrossRef] [PubMed]
- Wenner, H.A.; Macasaet, F.D.; Kamitsuka, P.S.; Kidd, P. Monkey pox. I. Clinical, virologic and immunologic studies. Am. J. Epidemiol. 1968, 87, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Sbrana, E.; Xiao, S.Y.; Newman, P.C.; Tesh, R.B. Comparative pathology of North American and central African strains of monkeypox virus in a ground squirrel model of the disease. Am. J. Trop. Med. Hyg. 2007, 76, 155–164. [Google Scholar] [PubMed]
- Tesh, R.B.; Watts, D.M.; Sbrana, E.; Siirin, M.; Popov, V.L.; Xiao, S.Y. Experimental infection of ground squirrels (Spermophilus tridecemlineatus) with monkeypox virus. Emerg. Infect. Dis. 2004, 10, 1563–1567. [Google Scholar] [CrossRef] [PubMed]
- Guarner, J.; Johnson, B.J.; Paddock, C.D.; Shieh, W.J.; Goldsmith, C.S.; Reynolds, M.G.; Damon, I.K.; Regnery, R.L.; Zaki, S.R. Monkeypox transmission and pathogenesis in prairie dogs. Emerg. Infect. Dis. 2004, 10, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.K.; Self, J.; Weiss, S.; Carroll, D.; Braden, Z.; Regnery, R.L.; Davidson, W.; Jordan, R.; Hruby, D.E.; Damon, I.K. Effective antiviral treatment of systemic orthopoxvirus disease: ST-246 treatment of prairie dogs infected with monkeypox. J. Virol. 2011, 85, 9176–9187. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.Y.; Sbrana, E.; Watts, D.M.; Siirin, M.; da Rosa, A.P.; Tesh, R.B. Experimental infection of prairie dogs with monkeypox virus. Emerg. Infect. Dis. 2005, 11, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.A.; Sagartz, J.E.; Huso, D.L.; Buller, R.M. Experimental infection of an African dormouse (Graphiurus kelleni) with monkeypox virus. Virology 2009, 383, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Americo, J.L.; Moss, B.; Earl, P.L. Identification of wild-derived inbred mouse strains highly susceptible to monkeypox virus infection for use as small animal models. J. Virol. 2010, 84, 8172–8180. [Google Scholar] [CrossRef] [PubMed]
- Hutson, C.L.; Abel, J.A.; Carroll, D.S.; Olson, V.A.; Braden, Z.H.; Hughes, C.M.; Dillon, M.; Hopkins, C.; Karem, K.L.; Damon, I.K.; et al. Comparison of West African and Congo Basin monkeypox viruses in BALB/c and C57BL/6 mice. PLoS ONE 2010, 5, e8912. [Google Scholar] [CrossRef] [PubMed]
- Marennikova, S.S.; Seluhina, E.M. Susceptibility of some rodent species to monkeypox virus, and course of the infection. Bull. World Health Organ. 1976, 53, 13–20. [Google Scholar] [PubMed]
- Osorio, J.E.; Iams, K.P.; Meteyer, C.U.; Rocke, T.E. Comparison of monkeypox viruses pathogenesis in mice by in vivo imaging. PLoS ONE 2009, 4, e6592. [Google Scholar]
- Shchelukhina, E.M.; Marennikova, S.S. Generalized monkeypox in orally infected rabbits and white mice. Vopr. Virusol. 1975, 6, 703–705. [Google Scholar]
- Stabenow, J.; Buller, R.M.; Schriewer, J.; West, C.; Sagartz, J.E.; Parker, S. A mouse model of lethal infection for evaluating prophylactics and therapeutics against Monkeypox virus. J. Virol. 2010, 84, 3909–3920. [Google Scholar] [PubMed]
- Buller, R.L.; Handley, L.; Parker, S. Development of prophylactics and therapeutics against the smallpox and monkeypox biothreat agents. In National Institute of Allergy and Infectious Disease: Frontiers in Research; St Georgiew, V., Wester, K., McGowan, J., Eds.; Humana Press: Clifton, NJ, USA, 2008; Volume 1, pp. 145–161. [Google Scholar]
- Esposito, J.J.; Knight, J.C. Orthopoxvirus DNA: A comparison of restriction profiles and maps. Virology 1985, 143, 230–251. [Google Scholar] [PubMed]
- Esposito, J.J.; Sammons, S.A.; Frace, A.M.; Osborne, J.D.; Olsen-Rasmussen, M.; Zhang, M.; Govil, D.; Damon, I.K.; Kline, R.; Laker, M.; et al. Genome sequence diversity and clues to the evolution of variola (smallpox) virus. Science 2006, 313, 807–812. [Google Scholar] [PubMed]
- Lourie, B.; Nakano, J.H.; Kemp, G.E.; Setzer, H.W. Isolation of poxvirus from an African rodent. J. Infect. Dis. 1975, 132, 677–681. [Google Scholar] [PubMed]
- Parker, S.; Schultz, D.A.; Meyer, H.; Buller, R.L. Smallpox and Monkeypox Viruses. In Encyclopedia of Virology; Mahy, B.W.J., Van Ragenmortel, N.H.V., Eds.; Elsevier: Frankfurt, Germany, 2008; Volume 3, pp. 639–644. [Google Scholar]
- Meraz, M.A.; White, J.M.; Sheehan, K.C.; Bach, E.A.; Rodig, S.J.; Dighe, A.S.; Kaplan, D.H.; Riley, J.K.; Greenlund, A.C.; Campbell, D.; et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 1996, 84, 431–442. [Google Scholar] [PubMed]
- Durbin, J.E.; Hackenmiller, R.; Simon, M.C.; Levy, D.E. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 1996, 84, 443–450. [Google Scholar] [PubMed]
- Parker, S.; Touchette, E.; Oberle, C.; Almond, M.; Robertson, A.; Trost, L.C.; Lampert, B.; Painter, G.; Buller, R.M. Efficacy of therapeutic intervention with an oral ether-lipid analogue of cidofovir (CMX001) in a lethal mousepox model. Antivir. Res. 2008, 77, 39–49. [Google Scholar] [PubMed]
- Moss, B.; Earl, P.L. Expression of proteins in mammalian cells using vaccinia cirus vectors. Current Protocols in Molecular Biology. In Overview of the Vaccinia Virus Expression System; Wiley: Hoboken, NJ, USA, 1998. [Google Scholar]
- Esteban, D.; Parker, S.; Schriewer, J.; Hartzler, H.; Buller, R.M. Mousepox, a small animal model of smallpox. Methods Mol. Biol. 2012, 890, 177–198. [Google Scholar] [PubMed]
- Wallace, G.D.; Buller, R.M. Kinetics of ectromelia virus (mousepox) transmission and clinical response in C57BL/6j, BALB/cByj and AKR/J inbred mice. Lab. Anim. Sci. 1985, 35, 41–46. [Google Scholar] [PubMed]
- Kermekchiev, M.B.; Kirilova, L.I.; Vail, E.E.; Barnes, W.M. Mutants of Taq DNA polymerase resistant to PCR inhibitors allow DNA amplification from whole blood and crude soil samples. Nucleic Acids Res. 2009, 37, e40. [Google Scholar] [CrossRef] [PubMed]
- Esteban, D.J.; Buller, R.M. Ectromelia virus: The causative agent of mousepox. J. Gen. Virol. 2005, 86, 2645–2659. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.; Siddiqui, A.M.; Oberle, C.; Hembrador, E.; Lanier, R.; Painter, G.; Robertson, A.; Buller, R.M. Mousepox in the C57BL/6 strain provides an improved model for evaluating anti-poxvirus therapies. Virology 2009, 385, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.; Siddiqui, A.M.; Painter, G.; Schriewer, J.; Buller, R.M. Ectromelia Virus Infections of Mice as a Model to Support the Licensure of Anti-Orthopoxvirus Therapeutics. Viruses 2010, 2, 1914. [Google Scholar] [CrossRef] [PubMed]
- Karupiah, G.; Buller, R.M.; Van Rooijen, N.; Duarte, C.J.; Chen, J. Different roles for CD4+ and CD8+ T lymphocytes and macrophage subsets in the control of a generalized virus infection. J. Virol. 1996, 70, 8301–8309. [Google Scholar] [PubMed]
- Karupiah, G. Type 1 and type 2 cytokines in antiviral defense. Vet. Immunol. Immunopathol. 1998, 63, 105–109. [Google Scholar] [CrossRef]
- Bradley, D.J.; Grainger, W.E. A poxvirus infection of gerbils in Uganda. Trans. R. Soc. Trop. Med. Hyg. 1971, 65, 26. [Google Scholar] [PubMed]
- Wilson, D.E.; Reeder, D.M. Mammal Species of the World. A Taxonomic and Gerographic Reference, 3rd ed.; Johns Hopkins University Press: Baltimore, MD, USA, 2005. [Google Scholar]
- Neyts, J.; De Clercq, E. Efficacy of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine for the treatment of lethal vaccinia virus infections in severe combined immune deficiency (SCID) mice. J. Med. Virol. 1993, 41, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Quenelle, D.C.; Collins, D.J.; Kern, E.R. Efficacy of multiple- or single-dose cidofovir against vaccinia and cowpox virus infections in mice. Antimicrob. Agents Chemother. 2003, 47, 3275–3280. [Google Scholar] [CrossRef] [PubMed]
- Smee, D.F.; Bailey, K.W.; Sidwell, R.W. Treatment of cowpox virus respiratory infections in mice with ribavirin as a single agent or followed sequentially by cidofovir. Antivir. Chem. Chemother. 2000, 11, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.; Martinez, M.; Smee, D.F.; Kefauver, D.; Thompson, E.; Huggins, J.W. Cidofovir protects mice against lethal aerosol or intranasal cowpox virus inoculation. J. Infect. Dis. 2000, 181, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.B. The cause of death in smallpox: An examination of the pathology record. Mil. Med. 2002, 167, 546–551. [Google Scholar] [PubMed]
- Nelson, J.B. The Behavior of Pox Viruses in the Respiratory Tract: I. The Response of Mice to the Nasal Instillation of Vaccinia Virus. J. Exp. Med. 1938, 68, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Smee, D.F.; Bailey, K.W.; Wong, M.H.; Sidwell, R.W. Effects of cidofovir on the pathogenesis of a lethal vaccinia virus respiratory infection in mice. Antivir. Res. 2001, 52, 55–62. [Google Scholar] [CrossRef]
- Martinez, M.J.; Bray, M.P.; Huggins, J.W. A mouse model of aerosol-transmitted orthopoxviral disease: Morphology of experimental aerosol-transmitted orthopoxviral disease in a cowpox virus-BALB/c mouse system. Arch. Pathol. Lab. Med. 2000, 124, 362–377. [Google Scholar] [PubMed]
- Saijo, M.; Ami, Y.; Suzaki, Y.; Nagata, N.; Iwata, N.; Hasegawa, H.; Iizuka, I.; Shiota, T.; Sakai, K.; Ogata, M.; et al. Virulence and pathophysiology of the Congo Basin and West African strains of monkeypox virus in non-human primates. J. Gen. Virol. 2009, 90, 2266–2271. [Google Scholar] [CrossRef] [PubMed]
- Zaucha, G.M.; Jahrling, P.B.; Geisbert, T.W.; Swearengen, J.R.; Hensley, L. The pathology of experimental aerosolized monkeypox virus infection in cynomolgus monkeys (Macaca fascicularis). Lab. Investig. 2001, 81, 1581–1600. [Google Scholar] [CrossRef] [PubMed]
- Schell, K. Studies on the innate resistance of mice to infection with mousepox. I. Resistance and antibody production. Aust. J. Exp. Biol. Med. Sci. 1960, 38, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Fenner, F. Studies in mousepox, infectious ectromelia of mice; quantitative investigations on the spread of virus through the host in actively and passively immunized animals. Aust. J. Exp. Biol. Med. Sci. 1949, 27, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Norbury, C.C.; Malide, D.; Gibbs, J.S.; Bennink, J.R.; Yewdell, J.W. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat. Immunol. 2002, 3, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Fenner, F. Studies in infectious ectromelia in mice; natural transmission; the portal of entry of the virus. Aust. J. Exp. Biol. Med. Sci. 1947, 25, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Chaudhri, G.; Panchanathan, V.; Buller, R.M.; van den Eertwegh, A.J.; Claassen, E.; Zhou, J.; de Chazal, R.; Laman, J.D.; Karupiah, G. Polarized type 1 cytokine response and cell-mediated immunity determine genetic resistance to mousepox. Proc. Natl. Acad. Sci. USA 2004, 101, 9057–9062. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunocompetent | |||||
| Species/Strain | Route 2 | Seroconvert 3 | Mortality (%) | Day of Death | Weight Loss (%) 5 |
| Gerbil | IN/FP | + | 0 | N/A 4 | - |
| Dormouse | IN/FP | − | 0 | N/A | - |
| A/Ncr | IN/FP | + | 0 | N/A | - |
| SKH-1 | IN/FP | + | 0 | N/A | - |
| C57BL/6 | IN/FP | + | 0 | N/A | - |
| CAST/EiJ | IN/FP | + | 0 | N/A | - |
| 129 | IN/FP | + | 0 | N/A | - |
| Immunodeficient | |||||
| Species/Strain | Route | Seroconvert | Mortality (%) | Day of Death | Weight Loss (%) 5 |
| 129 stat1−/− | IN/FP | + | 0 | N/A | - |
| C57BL/6 stat1−/− | IN/FP | + | 0 | N/A | - |
| SCID (BALB/c) | IN | N/A | 100 | 31 ± 3 | 34.5 |
| SCID (BALB/c) | FP | N/A | 100 | 51 ± 5 | 28.3 |
| SCID (SKH1) | FP | N/A | 50 | 52 ± 0 | 15.8 |
| Cage | Virus 2 | TATV Inoculation Route (T = 0) 2 | Mice Seroconversion (T = 28) 3 | ECTV Inoculation Route (T = 35) 4 | DOD Following ECTV Challenge 5 |
|---|---|---|---|---|---|
| 1 | Mock | FP | −−− | FP | 7,7,9 |
| 2 | Mock | IN | −−− | IN | 7,8,7 |
| 3 | TV | FP | +++ | FP | |
| 4 | TV-psoralen | FP | −−+ 6 | FP | 6,7,ND 6 |
| 5 | TV | IN | +++ | IN | |
| 6 | TV-psoralen | IN | −−− | IN | 8,8,9 |
| Index | Route | Contact | ECTV 2 | TATV 2 |
|---|---|---|---|---|
| 129 | FP | 129 | 4/4 | 0/4 |
| 129 stat1−/− | FP | 129 stat1−/− | 0/43 | 0/4 |
| C57BL/6 | FP | C57BL/6 | 4/4 | 0/4 |
| C57BL/6 stat1−/− | FP | C57BL/6 stat1−/− | 0/4 3 | 0/4 |
| SCID | IN | C57BL/6 | 0/4 3 | 4/4 7 |
| SCID | IN | C57BL/6 stat1−/− | 4/4 3,4 | 4/4 7 |
| SCID | IN | A/Ncr | 4/4 3,5 | 0/4 |
| Gerbil | FP | Gerbil | N/A 6 | 0/4 |
| Gerbil | IN | Gerbil | N/A | 0/4 |
| Virus | Diagnosis | Distribution 2 | Severity 3 | Number Presenting 4 | Comments |
|---|---|---|---|---|---|
| TATV | Inflammation, subacute | MF | 2 | 4 | Plantar metatarsal region |
| Inflammation, subacute | MF | 3 | 2 | Plantar metatarsal region | |
| ECTV | Inflammation, subacute | MF | 1 | 5 | Plantar metatarsal region |
| Inflammation, subacute | MF | 1 | 1 | Plantar metatarsal region and periosteal (distal tibia) |
| Cytokine | ECTV | TATV | |||||
|---|---|---|---|---|---|---|---|
| Cytokine | Mock | ↑ vs. Mock | ≈ vs. Mock | ↓ vs. Mock | ↑ vs. Mock | ≈ vs. Mock | ↓ vs. Mock |
| Eotaxin | 44 ± 4 | 125 ± 14 | 37 ± 4 | ||||
| GM-CSF | 9 ± 3 | 78 ± 11 | 4 ± 1 | ||||
| M-CSF | 20 ± 2 | 52 ± 4 | 30 ± 6 | ||||
| TNF-α | 5 ± 1 | 12 ± 1 | 6 ± 1 | ||||
| IL-5 | 0.3 ± 0 | 10 ± 2 | 0.1 ± 0 | ||||
| IL-13 | 43 ± 15 | 297 ± 39 | 15 ± 4 | ||||
| MIP-1α | 25 ± 4 | 44 ± 4 | 18 ± 3 | ||||
| MIP-1β | 48 ± 7 | 105 ± 11 | 46 ± 4 | ||||
| IFN-γ | 19 ± 5 | 81 ± 10 | 8 ± 2 | ||||
| IP-10 | 91 ± 17 | 1232 ± 170 | 124 ± 31 | ||||
| LIF | 6 ± 2 | 42 ± 5 | 9 ± 2 | ||||
| IL-12p70 | 3 ± 1 | 20 ± 4 | 1 ± 0 | ||||
| MCP-1 | 58 ± 8 | 709 ± 60 | 116 ± 21 | ||||
| MIG | 949 ± 252 | 6896 ± 758 | 726 ± 116 | ||||
| RANTES | 79 ± 6 | 110 ± 7 | 76 ± 8 | ||||
| G-CSF | 251 ± 43 | 241 ± 40 | 89 ± 8 | ||||
| VEGF | 16 ± 2 | 11 ± 2 | 6 ± 1 | ||||
| IL-10 | 11 ± 2 | 7 ± 1 | 3 ± 1 | ||||
| IL-12p40 | 230 ± 52 | 137 ± 18 | 64 ± 18 | ||||
| IL-1β | 43 ± 6 | 44 ± 6 | 25 ± 3 | ||||
| IL-15 | 112 ± 22 | 93 ± 17 | 36 ± 9 | ||||
| IL-17 | 3 ± 1 | 4 ± 1 | 1 ± 0 | ||||
| IL-6 | 14 ± 3 | 189 ± 20 | 29 ± 3 | ||||
| KC | 23 ± 2 | 286 ± 25 | 56 ± 10 | ||||
| IL-3 | 1 ± 0.1 | 7 ± 1 | 3 ± 0.4 | ||||
| IL-9 | 532 ± 74 | 160 ± 44 | 145 ± 36 | ||||
| IL-1α | 115 ± 16 | 228 ± 29 | 73.2 ± 7 | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, S.; Crump, R.; Hartzler, H.; Buller, R.M. Evaluation of Taterapox Virus in Small Animals. Viruses 2017, 9, 203. https://doi.org/10.3390/v9080203
Parker S, Crump R, Hartzler H, Buller RM. Evaluation of Taterapox Virus in Small Animals. Viruses. 2017; 9(8):203. https://doi.org/10.3390/v9080203
Chicago/Turabian StyleParker, Scott, Ryan Crump, Hollyce Hartzler, and R. Mark Buller. 2017. "Evaluation of Taterapox Virus in Small Animals" Viruses 9, no. 8: 203. https://doi.org/10.3390/v9080203
APA StyleParker, S., Crump, R., Hartzler, H., & Buller, R. M. (2017). Evaluation of Taterapox Virus in Small Animals. Viruses, 9(8), 203. https://doi.org/10.3390/v9080203