
Classification of Cowpox Viruses into Several Distinct Clades and Identification of a Novel Lineage


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection and Isolation of CPXV Strains
2.2. High-Throughput Sequencing
2.3. De Novo Assembly and Genome Annotation
2.4. Accession Numbers
2.5. Phylogenetic Analysis
2.6. Geographic Analysis
3. Results
3.1. Novel CPXV Isolates
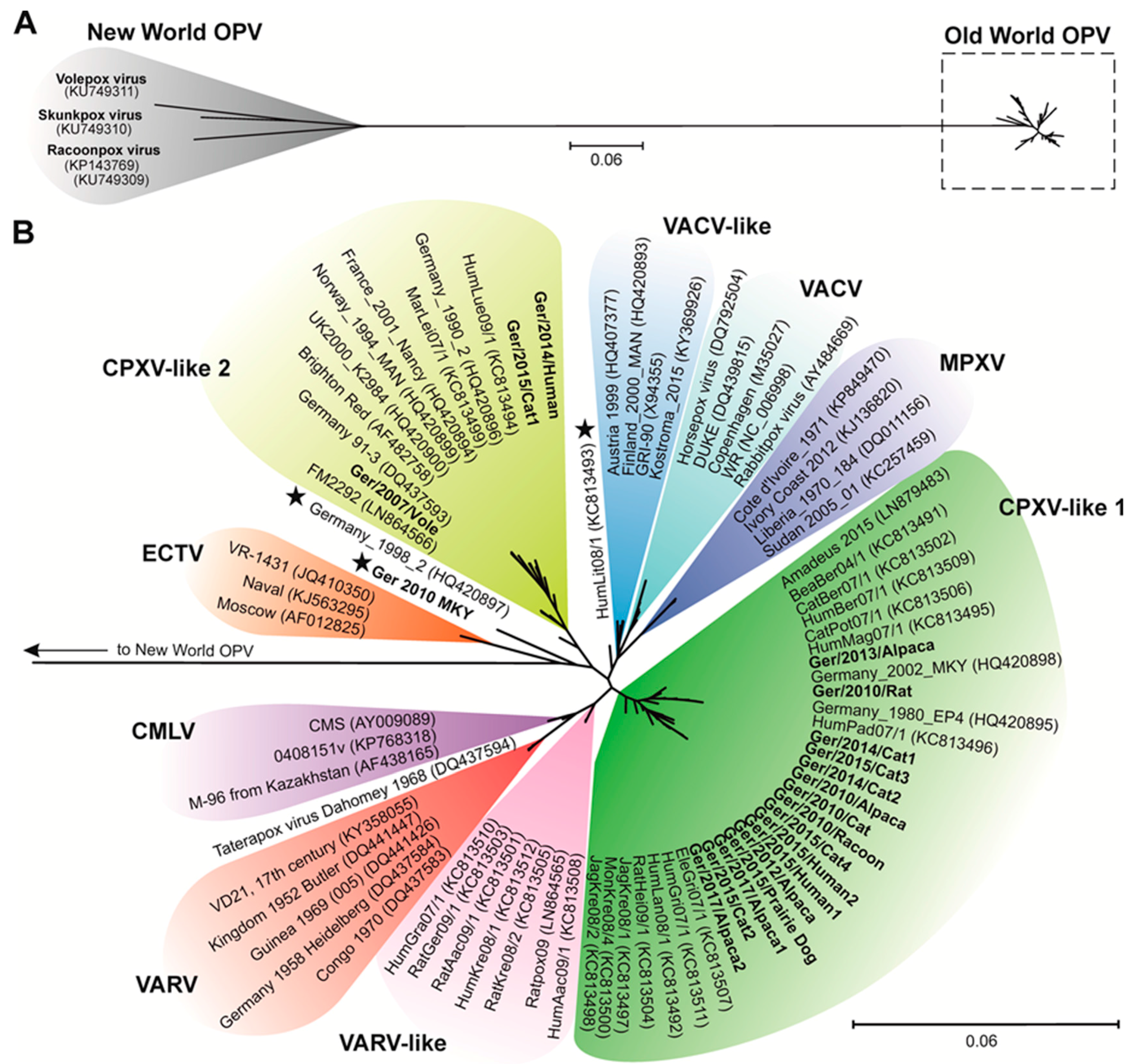
3.2. CPXV Phylogeny
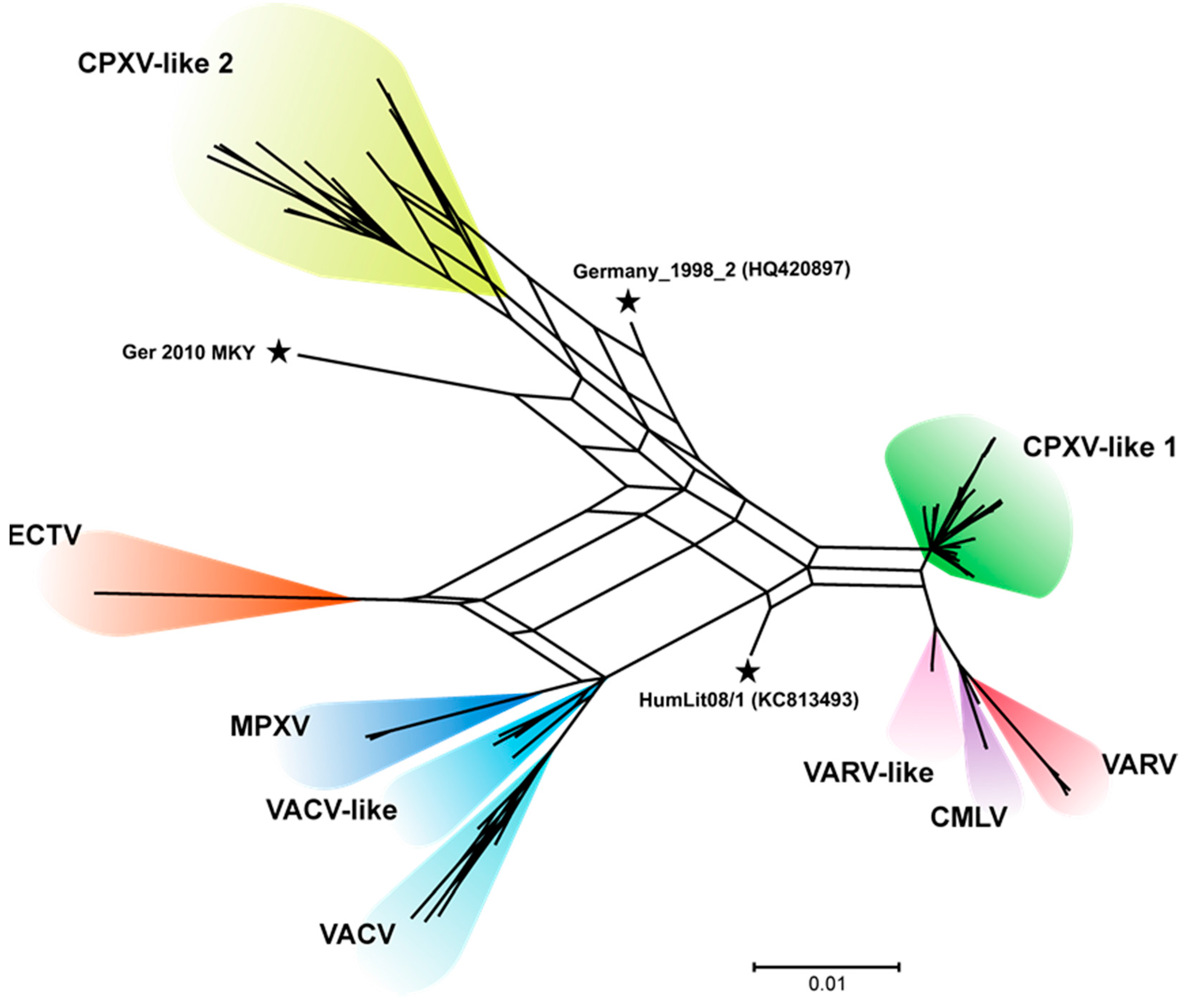
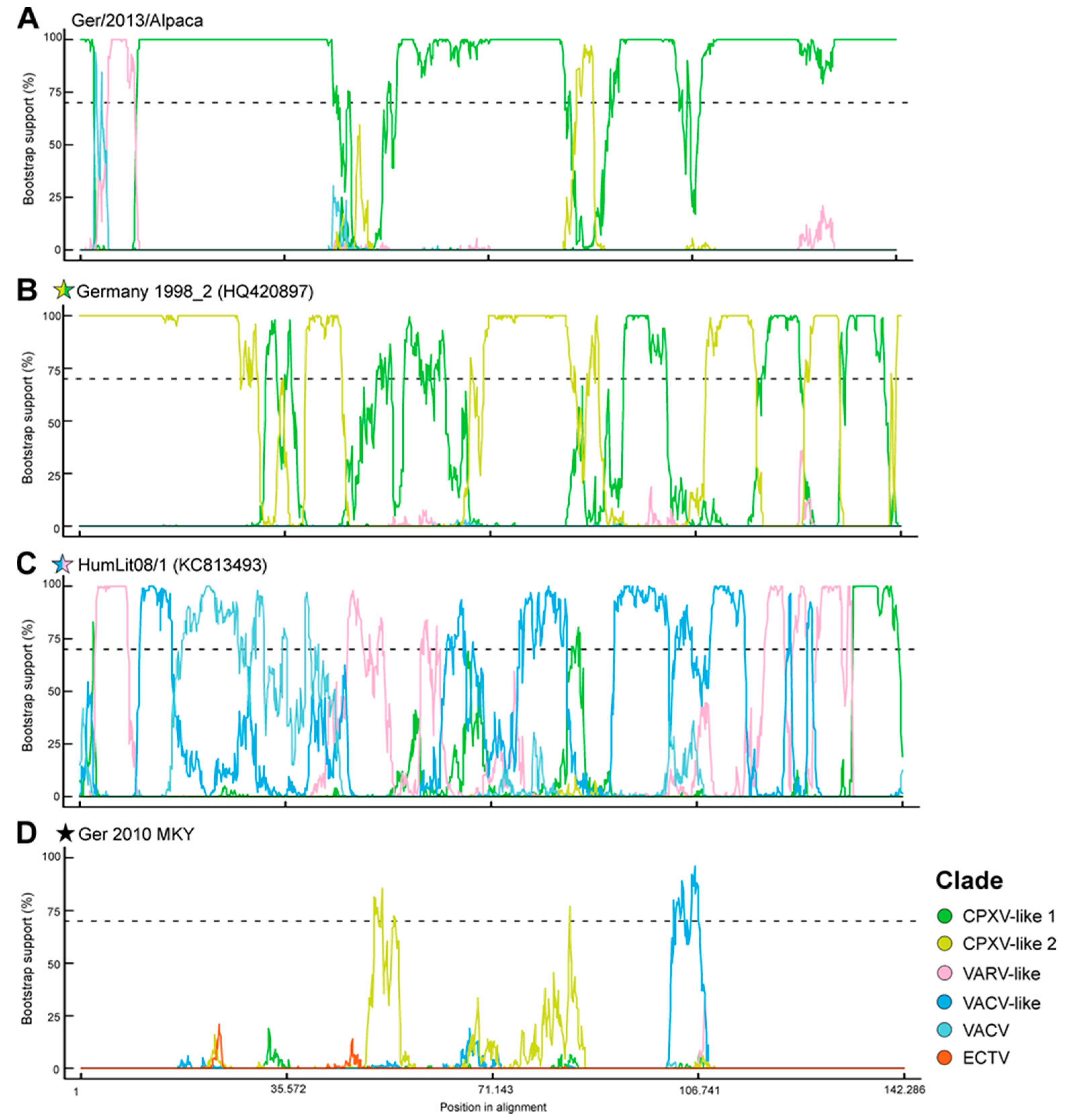
3.3. CPXV Consensus Network and Bootscan Analysis
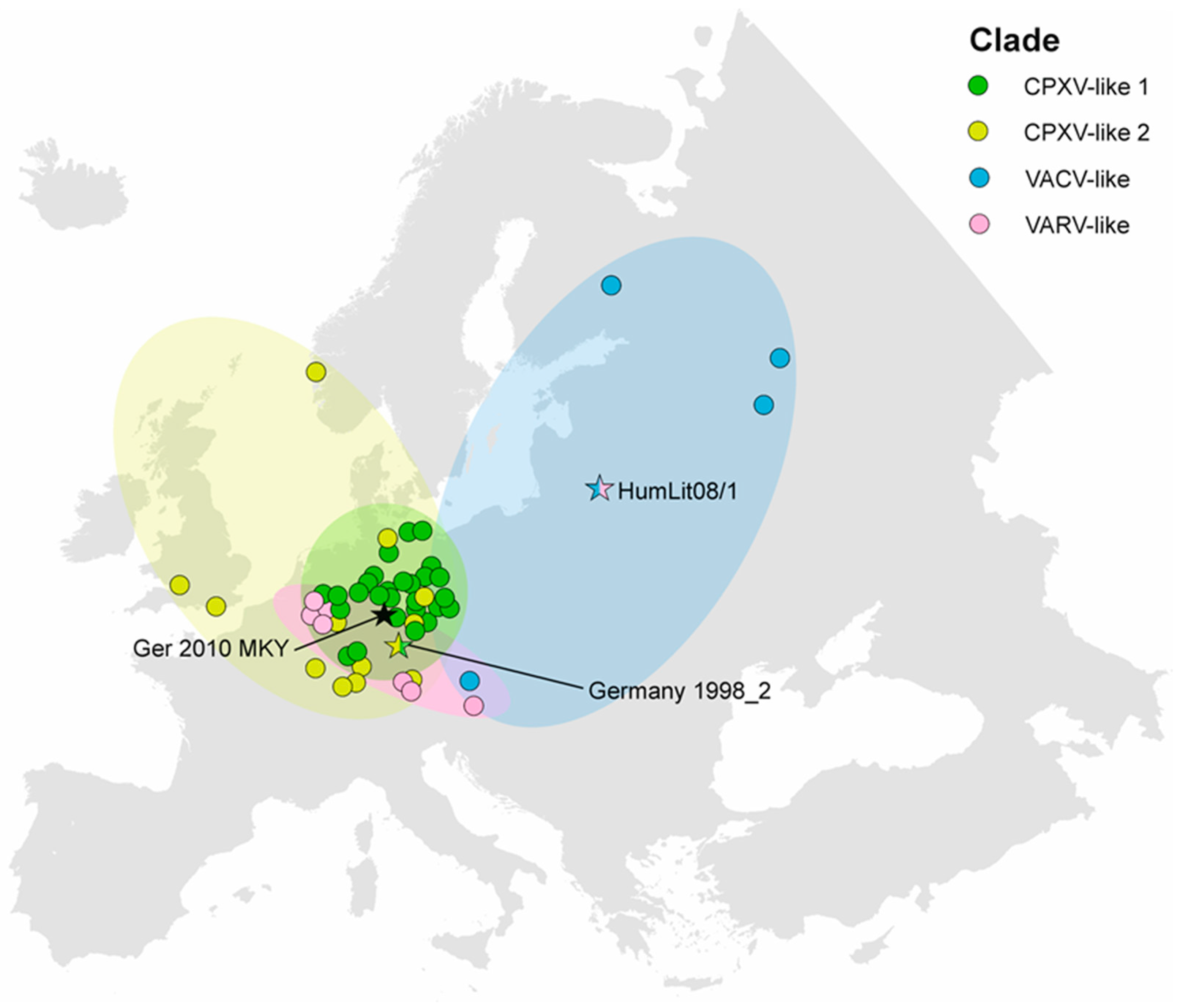
3.4. CPXV Geographic Distribution
4. Discussion
4.1. Phylogenetic Analysis
4.2. Geographic Distribution of CPXV Clades
4.2.1. CPXV Situation in Germany
4.2.2. CPXV Situation in Europe
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- International Committee on Taxonomy of Viruses (ICTV). In Virus Taxonomy: 2015 Release; International Committee on Taxonomy of Viruses, 2015.
- Sanchez-Sampedro, L.; Perdiguero, B.; Mejias-Perez, E.; Garcia-Arriaza, J.; Di Pilato, M.; Esteban, M. The evolution of poxvirus vaccines. Viruses 2015, 7, 1726–1803. [Google Scholar] [CrossRef] [PubMed]
- Downie, A.W. A study of the lesions produced experimentally by cowpox virus. J. Pathol. Bacteriol. 1939, 48, 361–379. [Google Scholar] [CrossRef]
- Archard, L.C.; Mackett, M. Restriction endonuclease analysis of red cowpox virus and its white pock variant. J. Gen. Virol. 1979, 45, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Chantrey, J.; Meyer, H.; Baxby, D.; Begon, M.; Bown, K.J.; Hazel, S.M.; Jones, T.; Montgomery, W.I.; Bennett, M. Cowpox: Reservoir hosts and geographic range. Epidemiol. Infect. 1999, 122, 455–460. [Google Scholar] [CrossRef] [PubMed]
- McFadden, G. Poxvirus tropism. Nat. Rev. Microbiol. 2005, 3, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Vorou, R.M.; Papavassiliou, V.G.; Pierroutsakos, I.N. Cowpox virus infection: An emerging health threat. Curr. Opin. Infect. Dis. 2008, 21, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Sardy, M.; Glos, K.; Korting, H.C.; Ruzicka, T.; Wollenberg, A. The munich outbreak of cutaneous cowpox infection: Transmission by infected pet rats. Acta Derm-Venereol. 2012, 92, 126–131. [Google Scholar] [PubMed]
- Becker, C.; Kurth, A.; Hessler, F.; Kramp, H.; Gokel, M.; Hoffmann, R.; Kuczka, A.; Nitsche, A. Cowpox virus infection in pet rat owners: Not always immediately recognized. Dtsch. Arztebl. Int. 2009, 106, 329–334. [Google Scholar] [PubMed]
- Bonnekoh, B.; Falk, K.; Reckling, K.F.; Kenklies, S.; Nitsche, A.; Ghebremedhin, B.; Pokrywka, A.; Franke, I.; Thriene, B.; Konig, W.; et al. Cowpox infection transmitted from a domestic cat. J. Dtsch. Dermatol. Ges. 2008, 6, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Switaj, K.; Kajfasz, P.; Kurth, A.; Nitsche, A. Cowpox after a cat scratch—Case report from poland. Ann. Agric. Environ. Med. 2015, 22, 456–458. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, C.J.; Littmann, M.; Lobermann, M.; Meyer, H.; Petschaelis, A.; Reisinger, E.C. Human cowpox virus infection acquired from a circus elephant in Germany. Int. J. Infect. Dis. 2010, 14, e338–e340. [Google Scholar] [CrossRef] [PubMed]
- Kurth, A.; Wibbelt, G.; Gerber, H.P.; Petschaelis, A.; Pauli, G.; Nitsche, A. Rat-to-elephant-to-human transmission of cowpox virus. Emerg. Infect. Dis. 2008, 14, 670–671. [Google Scholar] [CrossRef] [PubMed]
- Kurth, A.; Straube, M.; Kuczka, A.; Dunsche, A.J.; Meyer, H.; Nitsche, A. Cowpox virus outbreak in banded mongooses (Mungos mungo) and jaguarundis (Herpailurus yagouaroundi) with a time-delayed infection to humans. PLoS ONE 2009, 4, e6883. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, P.; Zange, S.; Ibrahim, S.; Zoeller, G.; Herbstreit, F.; Meyer, H. Generalized cowpox virus infection in a patient with HIV, Germany, 2012. Emerg. Infect. Dis. 2016, 22, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, P.M.; Holopainen, J.M.; Hemmila, H.; Piiparinen, H.; Sironen, T.; Kivela, T.; Virtanen, J.; Niemimaa, J.; Nikkari, S.; Jarvinen, A.; et al. Severe ocular cowpox in a human, Finland. Emerg. Infect. Dis. 2015, 21, 2261–2263. [Google Scholar] [CrossRef] [PubMed]
- Czerny, C.P.; Eis-Hubinger, A.M.; Mayr, A.; Schneweis, K.E.; Pfeiff, B. Animal poxviruses transmitted from cat to man: Current event with lethal end. Zentralbl Veterinarmed Reihe B 1991, 38, 421–431. [Google Scholar] [CrossRef]
- Haase, O.; Moser, A.; Rose, C.; Kurth, A.; Zillikens, D.; Schmidt, E. Generalized cowpox infection in a patient with darier disease. Br. J. Dermatol. 2011, 164, 1116–1118. [Google Scholar] [CrossRef] [PubMed]
- Esposito, J.J.; Knight, J.C. Orthopoxvirus DNA: A comparison of restriction profiles and maps. Virology 1985, 143, 230–251. [Google Scholar] [CrossRef]
- Gubser, C.; Hue, S.; Kellam, P.; Smith, G.L. Poxvirus genomes: A phylogenetic analysis. J. Gen. Virol. 2004, 85, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxviridae: The viruses and their replication. In Field's Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 2001; pp. 2849–2883. [Google Scholar]
- Patel, D.D.; Pickup, D.J.; Joklik, W.K. Isolation of cowpox virus a-type inclusions and characterization of their major protein component. Virology 1986, 149, 174–189. [Google Scholar] [CrossRef]
- Pfeffer, M.; Meyer, H. Poxvirus diagnostics. In Poxviruses; Mercer, A.A., Schmidt, A., Weber, O., Eds.; Birkhäuser Verlag: Basel, Switzerland, 2007; pp. 355–374. [Google Scholar]
- Kurth, A.; Nitsche, A. Fast and reliable diagnostic methods for the detection of human poxvirus infections. Future Virol. 2007, 2, 467–479. [Google Scholar] [CrossRef]
- Carroll, D.S.; Emerson, G.L.; Li, Y.; Sammons, S.; Olson, V.; Frace, M.; Nakazawa, Y.; Czerny, C.P.; Tryland, M.; Kolodziejek, J.; et al. Chasing jenner’s vaccine: Revisiting cowpox virus classification. PLoS ONE 2011, 6, e23086. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, P.W.; Radonic, A.; Kurth, A.; Nitsche, A. Genome-wide comparison of cowpox viruses reveals a new clade related to variola virus. PLoS ONE 2013, 8, e79953. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Franke, A.; Jenckel, M.; Tamosiunaite, A.; Schluckebier, J.; Granzow, H.; Hoffmann, B.; Fischer, S.; Ulrich, R.G.; Höper, D.; et al. Out of the reservoir: Phenotypic and genotypic characterization of a novel cowpox virus isolated from a common vole. J. Virol. 2015, 89, 10959–10969. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, A.; Ellerbrok, H.; Pauli, G. Detection of orthopoxvirus DNA by real-time PCR and identification of variola virus DNA by melting analysis. J. Clin. Microbiol. 2004, 42, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Miernik, B.; Casetti, F.; Panning, M.; Huzly, D.; Meyer, H.; Technau-Hafsi, K. Multilocular facial necrosis in a young boy: A quiz. Acta Derm. Venereol. 2017, 97, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Kalthoff, D.; Bock, W.I.; Huhn, F.; Beer, M.; Hoffmann, B. Fatal cowpox virus infection in cotton-top tamarins (Saguinus oedipus) in Germany. Vector Borne Zoonotic Dis. 2014, 14, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Holland, B.; Moulton, V. Consensus networks: A method for visualising incompatibilities in collections of trees. In Proceedings of the 3rd International Workshop Algorithms Bioinformatics (WABI), Budapest, Hungary, 15–20 September 2003; pp. 165–176. [Google Scholar]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Essbauer, S.; Pfeffer, M.; Meyer, H. Zoonotic poxviruses. Vet. Microbiol. 2010, 140, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; Kershaw, O.; Jenckel, M.; König, L.; Beer, M.; Hoffmann, B.; Hoffmann, D. Fatal cowpox virus infection in an aborted foal. Vector Borne Zoonotic Dis. 2016, 16, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Kolb, A.W.; Ane, C.; Brandt, C.R. Using HSV-1 genome phylogenetics to track past human migrations. PLoS ONE 2013, 8, e76267. [Google Scholar] [CrossRef] [PubMed]
- Gershon, P.D.; Kitching, R.P.; Hammond, J.M.; Black, D.N. Poxvirus genetic recombination during natural virus transmission. J. Gen. Virol. 1989, 70, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Okeke, M.I.; Hansen, H.; Traavik, T. A naturally occurring cowpox virus with an ectromelia virus A-type inclusion protein gene displays atypical A-type inclusions. Infect. Genet. Evol. 2012, 12, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.; Okeke, M.I.; Nilssen, O.; Traavik, T. Recombinant viruses obtained from co-infection in vitro with a live vaccinia-vectored influenza vaccine and a naturally occurring cowpox virus display different plaque phenotypes and loss of the transgene. Vaccine 2004, 23, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Chernos, V.I.; Antonova, T.P.; Senkevich, T.G. Recombinants between vaccinia and ectromelia viruses bearing the specific pathogenicity markers of both parents. J. Gen. Virol. 1985, 66, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.A. High-frequency homologous recombination in vaccinia virus DNA. J. Virol. 1987, 61, 1788–1795. [Google Scholar] [PubMed]
- Fathi, Z.; Dyster, L.M.; Seto, J.; Condit, R.C.; Niles, E.G. Intragenic and intergenic recombination between temperature-sensitive mutants of vaccinia virus. J. Gen. Virol. 1991, 72, 2733–2737. [Google Scholar] [CrossRef] [PubMed]
- Esposito, J.J.; Sammons, S.A.; Frace, A.M.; Osborne, J.D.; Olsen-Rasmussen, M.; Zhang, M.; Govil, D.; Damon, I.K.; Kline, R.; Laker, M.; et al. Genome sequence diversity and clues to the evolution of variola (smallpox) virus. Science 2006, 313, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Ninove, L.; Domart, Y.; Vervel, C.; Voinot, C.; Salez, N.; Raoult, D.; Meyer, H.; Capek, I.; Zandotti, C.; Charrel, R.N. Cowpox virus transmission from pet rats to humans, France. Emerg. Infect. Dis. 2009, 15, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Campe, H.; Zimmermann, P.; Glos, K.; Bayer, M.; Bergemann, H.; Dreweck, C.; Graf, P.; Weber, B.K.; Meyer, H.; Buttner, M.; et al. Cowpox virus transmission from pet rats to humans, Germany. Emerg. Infect. Dis. 2009, 15, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Ducournau, C.; Ferrier-Rembert, A.; Ferraris, O.; Joffre, A.; Favier, A.L.; Flusin, O.; Van Cauteren, D.; Kecir, K.; Auburtin, B.; Vedy, S.; et al. Concomitant human infections with 2 cowpox virus strains in related cases, France, 2011. Emerg. Infect. Dis. 2013, 19, 1996–1999. [Google Scholar] [CrossRef] [PubMed]
- Goerigk, D.; Theuß, T.; Pfeffer, M.; Konrath, A.; Kalthoff, D.; Woll, D.; Vahlenkamp, T.W.; Beer, M.; Starke, A. Kuhpockenvirusinfektion bei einem alpaka (Vicugna pacos)—Klinische Symptomatik, Diagnostik und pathologische befunde. Tierärztliche Praxis Großtiere 2014, 42, 169–177. [Google Scholar]
- Vora, N.M.; Li, Y.; Geleishvili, M.; Emerson, G.L.; Khmaladze, E.; Maghlakelidze, G.; Navdarashvili, A.; Zakhashvili, K.; Kokhreidze, M.; Endeladze, M.; et al. Human infection with a zoonotic orthopoxvirus in the country of georgia. N. Engl. J. Med. 2015, 372, 1223–1230. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Sampling Place | Year | Host | Clinical Description |
|---|---|---|---|---|
| Ger/2007/Vole | Rottweil | 2007 | Common vole | No clinical signs |
| Ger/2010/Alpaca | Oberwiesenthal | 2010 | Alpaca | Diseased |
| Ger 2010 MKY | Bad Liebenstein | 2010 | Cotton-top tamarin | Fatal generalization |
| Ger/2010/Cat | Nordhausen | 2010 | Cat | Fatal generalization |
| Ger/2010/Raccoon | Ellrich | 2010 | Raccoon | Fatal generalization |
| Ger/2010/Rat | Hannover | 2010 | Rat | Diseased |
| Ger/2012/Alpaca | Rositz | 2012 | Alpaca | Fatal generalization |
| Ger/2013/Alpaca | Zernitz | 2013 | Alpaca | Fatal generalization |
| Ger/2014/Cat1 | Bleckede | 2014 | Cat | Fatal generalization |
| Ger/2014/Cat2 | Nordhausen | 2014 | Cat | Fatal generalization |
| Ger/2014/Human | Freiburg | 2014 | Human | Local lesions |
| Ger/2015/Cat1 | Vogtlandkreis 1 | 2015 | Cat | Fatal generalization |
| Ger/2015/Cat2 | Rostock | 2015 | Cat | Local lesions |
| Ger/2015/Cat3 | Vogtlandkreis 1 | 2015 | Cat | Fatal generalization |
| Ger/2015/Cat4 | Hengelbach | 2015 | Cat | Local lesions |
| Ger/2015/Prairie-dog | Dresden | 2015 | Prairie dog | Local lesions |
| Ger/2015/Human1 | Leipzig | 2015 | Human | Cervical local lesion |
| Ger/2015/Human2 | Leipzig | 2015 | Human | Local lesion |
| Ger/2017/Alpaca1 | Brand-Erbisdorf | 2017 | Alpaca | Fatal generalization |
| Ger/2017/Alpaca2 | Merzdorf | 2017 | Alpaca | Fatal generalization |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franke, A.; Pfaff, F.; Jenckel, M.; Hoffmann, B.; Höper, D.; Antwerpen, M.; Meyer, H.; Beer, M.; Hoffmann, D. Classification of Cowpox Viruses into Several Distinct Clades and Identification of a Novel Lineage. Viruses 2017, 9, 142. https://doi.org/10.3390/v9060142
Franke A, Pfaff F, Jenckel M, Hoffmann B, Höper D, Antwerpen M, Meyer H, Beer M, Hoffmann D. Classification of Cowpox Viruses into Several Distinct Clades and Identification of a Novel Lineage. Viruses. 2017; 9(6):142. https://doi.org/10.3390/v9060142
Chicago/Turabian StyleFranke, Annika, Florian Pfaff, Maria Jenckel, Bernd Hoffmann, Dirk Höper, Markus Antwerpen, Hermann Meyer, Martin Beer, and Donata Hoffmann. 2017. "Classification of Cowpox Viruses into Several Distinct Clades and Identification of a Novel Lineage" Viruses 9, no. 6: 142. https://doi.org/10.3390/v9060142
APA StyleFranke, A., Pfaff, F., Jenckel, M., Hoffmann, B., Höper, D., Antwerpen, M., Meyer, H., Beer, M., & Hoffmann, D. (2017). Classification of Cowpox Viruses into Several Distinct Clades and Identification of a Novel Lineage. Viruses, 9(6), 142. https://doi.org/10.3390/v9060142