
Cowpox virus: What’s in a Name?

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culturing, Sequencing, Assembly, and Annotation
2.2. Phylogenetic Analysis
2.3. Species Delimitation and Species Tree Analyses
2.4. Topology, Dataset Heterogeneity, and Recombination Testing
3. Results
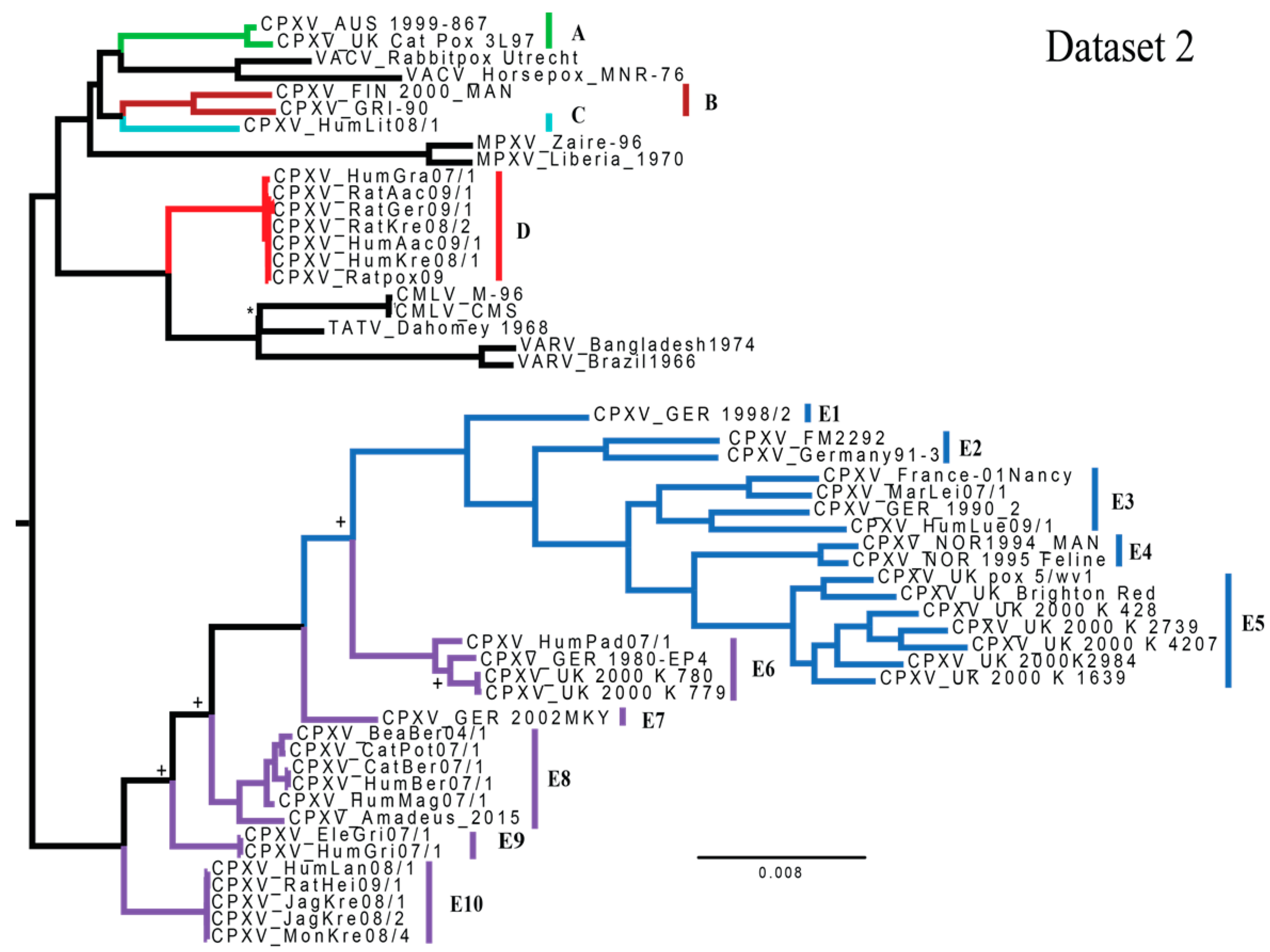
3.1. Phylogenetic Analyses
3.2. Species Delimitation and Species Tree Analyses
3.3. Topology, Dataset Heterogeneity, and Recombination Testing
4. Discussion
4.1. Phylogenetics and Species Delimitation
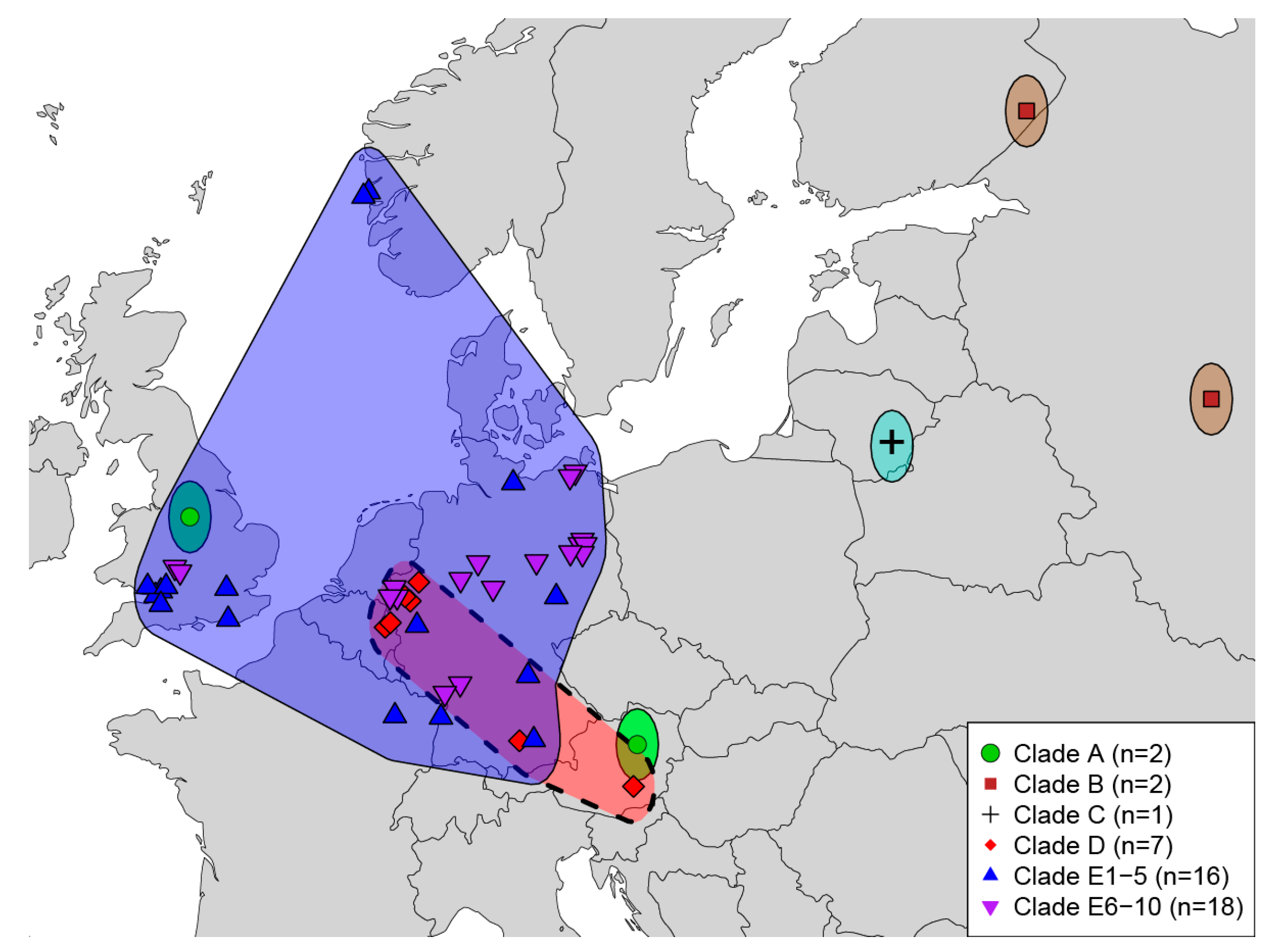
4.2. Geographic Sampling
4.3. Conclusions and Future Work
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scheffer, T.H. Habits and economic status of the pocket gophers. USDA Tech. Bull. 1931, 224, 26. [Google Scholar]
- Hyngstrom, S.; Timm, R.; Larson, G. Prevention and Control of Wildlife Damage; University of Nebraska Cooperative Extension: Lincoln, NE, USA, 1994; Volumes 1 and 2. [Google Scholar]
- International Congress on Taxonomy of Viruses Home Page. Available online: http://www.ictvonline.org (accessed on 5 May 2017).
- Adams, M.; Lefkowitz, E.; King, A.; Carstens, E. Recently agreed changes to the international code of virus classification and nomenclature. Arch. Virol. 2013, 158, 2633–2639. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.T. Defining viral species: Making taxonomy useful. Virol. J. 2014, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Mercer, A.; Schmidt, A.; Weber, O. Poxviruses; Springer Science & Business Media: Berlin, Germany, 2007. [Google Scholar]
- King, A.M.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Fenner, F.; Henderson, D.; Arita, I.; Jezek, Z.; Ladnyi, I. Smallpox and Its Eradication; World Health Organization: Geneva, Switzerland, 1988. [Google Scholar]
- Jenner, E. An Inquiry into the Causes and Effects of the Variolae Vaccinae: A Disease Discovered in Some of the Western Counties of England, Particularly Gloucestershire, and Known by the Name of the Cow Pox; Printed for the Author by Sampson Low, Berwick Street; SOHO: London, UK, 1798; Volume 7, p. 42. [Google Scholar]
- Jenner, E. Further Observations on the Variolæ Vaccinæ, or Cow–Pox; Printed for the Author by Sampson Low, Berwick Street; SOHO: London, UK, 1799; Volume 7. [Google Scholar]
- Davies, J.T.; Janes, L.; Downie, A. Cowpox infection in farmworkers. Lancet 1938, 232, 1534–1538. [Google Scholar] [CrossRef]
- Wildy, P. Classification and Nomenclature of Viruses: First Report of the International Committee on Nomenclature of Viruses; Karger: Basel, Switzerland, 1971; Volume 5. [Google Scholar]
- International Congress on Taxonomy of Viruses Taxonomy Release Page. Available online: Http://www.Ictvonline.Org/virustaxonomy.Asp (accessed 5 May 2017).
- Baxby, D. Laboratory characteristics of British and Dutch strains of Cowpox virus. Zoonoses Public Health 1975, 22, 480–487. [Google Scholar] [CrossRef]
- Baxby, D.; Ashton, D.; Jones, D.; Thomsett, L. An outbreak of cowpox in captive cheetahs: Virological and epidemiological studies. J. Hyg. 1982, 89, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Marennikova, S.S.; Maltseva, N.N.; Korneeva, V.I.; Garanina, N.M. Outbreak of pox disease among carnivora (Felidae) and edentata. J. Infect. Dis. 1977, 135, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Gehring, H.; Mahnel, H.; Mayer, H. Elephant pox. J. Vet. Med. Ser. B 1972, 19, 258–261. [Google Scholar]
- Essbauer, S.; Meyer, H. Genus orthopoxvirus: Cowpox virus. In Poxviruses; Springer: Berlin, Germany, 2007; pp. 75–88. [Google Scholar]
- Fenner, F.; Wittek, R.; Dumbell, K. The pathogenesis, pathology, and immunology of orthopoxvirus infections. In The Orthopoxviruses; Academic Press: San Diego, CA, USA, 1989; pp. 85–141. [Google Scholar]
- Stemmler, M.; Neubauer, H.; Meyer, H. Comparison of closely related orthopoxvirus isolates by random amplified polymorphic DNA and restriction fragment length polymorphism analysis. J. Vet. Med. Ser. B 2001, 48, 647–654. [Google Scholar] [CrossRef]
- Meyer, H.; Schay, C.; Mahnel, H.; Pfeffer, M. Characterization of orthopoxviruses isolated from man and animals in Germany. Arch. Virol. 1999, 144, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Baxby, D. Is cowpox misnamed? A review of 10 human cases. Br. Med. J. 1977, 1, 1379–1381. [Google Scholar] [CrossRef] [PubMed]
- Crouch, A.; Baxby, D.; McCracken, C.; Gaskell, R.; Bennett, M. Serological evidence for the reservoir hosts of Cowpox virus in British wildlife. Epidemiol. Infect. 1995, 115, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Ropp, S.L.; Jin, Q.; Knight, J.C.; Massung, R.F.; Esposito, J.J. PCR strategy for identification and differentiation of small pox and other orthopoxviruses. J. Clin. Microbiol. 1995, 33, 2069–2076. [Google Scholar] [PubMed]
- Pelkonen, P.M.; Tarvainen, K.; Hynninen, A.; Kallio, E.R.K.; Henttonen, H.; Palva, A.; Vaheri, A.; Vapalahti, O. Cowpox with severe generalized eruption, Finland. Emerg. Infect. Dis. 2003, 9, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Gubser, C.; Hue, S.; Kellam, P.; Smith, G.L. Poxvirus genomes: A phylogenetic analysis. J. Gen. Virol. 2004, 85, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D.S.; Emerson, G.L.; Li, Y.; Sammons, S.; Olson, V.; Frace, M.; Nakazawa, Y.; Czerny, C.P.; Tryland, M.; Kolodziejek, J. Chasing jenner’s vaccine: Revisiting Cowpox virus classification. PLoS ONE 2011, 6, e23086. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, P.W.; Radonić, A.; Kurth, A.; Nitsche, A. Genome-wide comparison of Cowpox viruses reveals a new clade related to variola virus. PLoS ONE 2013, 8, e79953. [Google Scholar]
- Hatcher, E.L.; Hendrickson, R.C.; Lefkowitz, E.J. Identification of nucleotide-level changes impacting gene content and genome evolution in orthopoxviruses. J. Virol. 2014, 88, 13651–13668. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, R.C.; Wang, C.; Hatcher, E.L.; Lefkowitz, E.J. Orthopoxvirus genome evolution: The role of gene loss. Viruses 2010, 2, 1933–1967. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; Kershaw, O.; Jenckel, M.; König, L.; Beer, M.; Hoffmann, B.; Hoffmann, D. Fatal Cowpox virus infection in an aborted foal. Vector-Borne Zoonotic Dis. 2016, 16, 431–433. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Adams, M.; Carstens, E.; Lefkowitz, E. The international code of virus classification and nomenclature. In Virus Taxonomy—Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier/Academic Press: London, UK, 2011; pp. 1273–1277. [Google Scholar]
- Kuhn, J.H.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Johnson, K.M.; Kawaoka, Y.; Lipkin, W.I.; Negredo, A.I.; Netesov, S.V.; Nichol, S.T. Proposal for a revised taxonomy of the family filoviridae: Classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 2010, 155, 2083–2103. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.; Martin, D.P.; Brown, J.K.; Navas-Castillo, J.; Moriones, E.; Zerbini, F.M.; Rivera-Bustamante, R.; Malathi, V.; Briddon, R.W.; Varsani, A. A genome-wide pairwise-identity-based proposal for the classification of viruses in the genus Mastrevirus (family Geminiviridae). Arch. Virol. 2013, 158, 1411–1424. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Berger, P.; Morales, F.; Myers, J.; Silbernagel, M.; Barnett, O. Taxonomy and classification of legume-infecting potyviruses. A proposal from the Potyviridae Study Group of the Plant Virus Subcommittee of ICTV. Arch. Virol. 1994, 139, 231–235. [Google Scholar]
- Hebert, P.D.; Stoeckle, M.Y.; Zemlak, T.S.; Francis, C.M. Identification of birds through DNA barcodes. PLoS Biol. 2004, 2, e312. [Google Scholar] [CrossRef] [PubMed]
- Leliaert, F.; Verbruggen, H.; Vanormelingen, P.; Steen, F.; López-Bautista, J.M.; Zuccarello, G.C.; De Clerck, O. DNA-based species delimitation in algae. Eur. J. Phycol. 2014, 49, 179–196. [Google Scholar] [CrossRef]
- Peterson, A.T.; Holder, M.T. Phylogenetic assessment of filoviruses: How many lineages of Marburg virus? Ecol. Evol. 2012, 2, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Yang, Z. Improved reversible jump algorithms for Bayesian species delimitation. Genetics 2013, 194, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Emerson, G.L.; Li, Y.; Frace, M.A.; Olsen-Rasmussen, M.A.; Khristova, M.L.; Govil, D.; Sammons, S.A.; Regnery, R.L.; Karem, K.L.; Damon, I.K. The phylogenetics and ecology of the orthopoxviruses endemic to North America. PLoS ONE 2009, 4, e7666. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. Varscan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef] [PubMed]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome annotation transfer utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov, S.N.; Totmenin, A.V.; Loparev, V.N.; Safronov, P.F.; Gutorov, V.V.; Chizhikov, V.E.; Knight, J.C.; Parsons, J.M.; Massung, R.F.; Esposito, J.J. Alastrim smallpox variola minor virus genome DNA sequences. Virology 2000, 266, 361–386. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Franke, A.; Jenckel, M.; Tamošiūnaitė, A.; Schluckebier, J.; Granzow, H.; Hoffmann, B.; Fischer, S.; Ulrich, R.G.; Höper, D.; et al. Out of the reservoir: Phenotypic and genotypic characterization of a novel Cowpox virus isolated from a common vole. J. Virol. 2015, 89, 10959–10969. [Google Scholar] [CrossRef] [PubMed]
- Esposito, J.J.; Sammons, S.A.; Frace, A.M.; Osborne, J.D.; Olsen-Rasmussen, M.; Zhang, M.; Govil, D.; Damon, I.K.; Kline, R.; Laker, M. Genome sequence diversity and clues to the evolution of variola (smallpox) virus. Science 2006, 313, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Pickup, D.J.; Bastia, D.; Stone, H.O.; Joklik, W.K. Sequence of terminal regions of Cowpox virus DNA: Arrangement of repeated and unique sequence elements. Proc. Natl. Acad. Sci. USA 1982, 79, 7112–7116. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.; Tulman, E.; Lu, Z.; Zsak, L.; Sandybaev, N.; Kerembekova, U.; Zaitsev, V.; Kutish, G.; Rock, D. The genome of camelpox virus. Virology 2002, 295, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gubser, C.; Smith, G.L. The sequence of camelpox virus shows it is most closely related to variola virus, the cause of smallpox. J. Gen. Virol. 2002, 83, 855–872. [Google Scholar] [CrossRef] [PubMed]
- Mavian, C.; López-Bueno, A.; Bryant, N.A.; Seeger, K.; Quail, M.A.; Harris, D.; Barrell, B.; Alcami, A. The genome sequence of ectromelia virus Naval and Cornell isolates from outbreaks in North America. Virology 2014, 462, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Danila, M.I.; Feng, Z.; Buller, R.M.L.; Wang, C.; Han, X.; Lefkowitz, E.J.; Upton, C. The genomic sequence of ectromelia virus, the causative agent of mousepox. Virology 2003, 317, 165–186. [Google Scholar] [CrossRef]
- Likos, A.M.; Sammons, S.A.; Olson, V.A.; Frace, A.M.; Li, Y.; Olsen-Rasmussen, M.; Davidson, W.; Galloway, R.; Khristova, M.L.; Reynolds, M.G. A tale of two clades: monkeypox viruses. J. Gen. Virol. 2005, 86, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov, S.; Totmenin, A.; Safronov, P.; Mikheev, M.; Gutorov, V.; Ryazankina, O.; Petrov, N.; Babkin, I.; Uvarova, E.; Sandakhchiev, L.; et al. Analysis of the monkeypox virus genome. Virology 2002, 297, 172–194. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, N.; Roper, R.; Feng, Z.; Hunter, A.; Danila, M.; Lefkowitz, E.; Buller, R.; Upton, C. Complete coding sequences of the rabbitpox virus genome. J. Gen. Virol. 2005, 86, 2969–2977. [Google Scholar] [CrossRef] [PubMed]
- Tulman, E.; Delhon, G.; Afonso, C.; Lu, Z.; Zsak, L.; Sandybaev, N.; Kerembekova, U.; Zaitsev, V.; Kutish, G.; Rock, D. Genome of horsepox virus. J. Virol. 2006, 80, 9244–9258. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with mafft. In Bioinformatics for DNA Sequence Analysis; Humana Press: New York, NY, USA, 2009; pp. 39–64. [Google Scholar]
- Bratke, K.A.; McLysaght, A. Identification of multiple independent horizontal gene transfers into poxviruses using a comparative genomics approach. BMC Evol. Biol. 2008, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A.; Holmes, E.C. Recombination in evolutionary genomics. Annu. Rev. Genet. 2002, 36, 75–97. [Google Scholar] [CrossRef] [PubMed]
- Nylander, J. MrModeltest, version 2; Evolutionary Biology Centre, Uppsala University: Uppsala, Sweden, 2004.
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony, version 4.0 b10. 2003.
- Springer, Y.P.; Hsu, C.H.; Werle, Z.R.; Olson, L.E.; Cooper, M.P.; Castrodale, L.J.; Fowler, N.; McCollum, A.M.; Goldsmith, C.S.; Emerson, G.L.; et al. Novel orthopoxvirus infection in an Alaska resident. Clin. Infect. Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Vora, N.M.; Li, Y.; Geleishvili, M.; Emerson, G.L.; Khmaladze, E.; Maghlakelidze, G.; Navdarashvili, A.; Zakhashvili, K.; Kokhreidze, M.; Endeladze, M. Human infection with a zoonotic orthopoxvirus in the country of Georgia. N. Engl. J. Med. 2015, 372, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Rannala, B. Bayesian species delimitation using multilocus sequence data. Proc. Natl. Acad. Sci. USA 2010, 107, 9264–9269. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, Y.; Mauldin, M.R.; Emerson, G.L.; Reynolds, M.G.; Lash, R.R.; Gao, J.; Zhao, H.; Li, Y.; Muyembe, J.J.; Kingebeni, P.M.; et al. A phylogeographic investigation of African monkeypox. Viruses 2015, 7, 2168–2184. [Google Scholar] [CrossRef] [PubMed]
- Massung, R.F.; Esposito, J.J.; Liu, L.-I.; Qi, J.; Utterback, T.R.; Knight, J.C.; Aubin, L.; Yuran, T.E.; Parsons, J.M.; Loparev, V.N. Potential virulence determinants in terminal regions of variola smallpox virus genome. Nature 1993, 366, 748. [Google Scholar] [CrossRef] [PubMed]
- Bruen, T.; Bruen, T. Phipack: PHI Test and Other Tests of Recombination; McGill University: Montreal, QC, Canada, 2005. [Google Scholar]
- Jakobsen, I.B.; Easteal, S. A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Comput. Appl. Biosci. 1996, 12, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed]
- Upton, C.; Slack, S.; Hunter, A.L.; Ehlers, A.; Roper, R.L. Poxvirus orthologous clusters: Toward defining the minimum essential poxvirus genome. J. Virol. 2003, 77, 7590–7600. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.; McFadden, G. Technical knockout: Understanding poxvirus pathogenesis by selectively deleting viral immunomodulatory genes. Cell. Microbiol. 2004, 6, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.; Nielsen, R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 2004, 167, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Pearl, D.K. Species trees from gene trees: Reconstructing Bayesian posterior distributions of a species phylogeny using estimated gene tree distributions. Syst. Biol. 2007, 56, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. The BPP program for species tree estimation and species delimitation. Curr. Zool. 2015, 61, 854–865. [Google Scholar] [CrossRef]
- Lanier, H.C.; Knowles, L.L. Is recombination a problem for species-tree analyses? Syst. Biol. 2012, 61, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Smithson, C.; Kampman, S.; Hetman, B.M.; Upton, C. Incongruencies in vaccinia virus phylogenetic trees. Computation 2014, 2, 182–198. [Google Scholar] [CrossRef]
- Okeke, M.I.; Hansen, H.; Traavik, T. A naturally occurring Cowpox virus with an ectromelia virus a-type inclusion protein gene displays atypical A-type inclusions. Infect. Genet. Evol. 2012, 12, 160–168. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Host | Year | Accession# | Location of Origin | Clade | Reference |
|---|---|---|---|---|---|---|---|
| CPXV | UK Cat Pox 3L97 | Cat | 1977 | KY549143 | UK | A | This paper |
| CPXV | AUS 1999-867 | Cat | 1999 | HQ407377 | Texing, Austria | [27] | |
| CPXV | GRI-90 | Human | 1990 | X94355 | Moscow, Russia | B | [45] |
| CPXV | FIN 2000_MAN | Human | 2000 | HQ420893 | Tohmajärvi, Finland | [27] | |
| CPXV | HumLit08/1 | Human | 2008 | KC813493 | Vilnius, Lithuania | C | [28] |
| CPXV | HumAac09/1 § | Human | 2009 | KC813508 | Aachen, Germany | D | [28] |
| CPXV | RatAac09/1 § | Rat | 2009 | KC813501 | Aachen, Germany | [28] | |
| CPXV | RatGer09/1 § | Rat | 2009 | KC813503 | Germering, Germany | [28] | |
| CPXV | HumKre08/1 § | Human | 2008 | KC813512 | Krefeld, Germany | [28] | |
| CPXV | RatKre08/2 § | Rat | 2008 | KC813505 | Krefeld, Germany | [28] | |
| CPXV | Ratpox09 § | Rat | 2009 | LN864565 | Marl, Germany | [46] | |
| CPXV | HumGra07/1 | Human | 2007 | KC813510 | Graz, Austria | [28] | |
| CPXV | GER 1998/2 | human | 1998 | HQ420897 | Eckental, Germany | E1 | [27] |
| CPXV | Germany 91-3 | Human | 1991 | DQ437593 | Munich, Germany | E2 | [47] |
| CPXV | FM2292 | Common Vole | 2011 | LN864566 | Baden-Wuerttemberg, Germany | [46] | |
| CPXV | MarLei07/1 | Mara | 2007 | KC813499 | Leipzig, Germany | E3 | [28] |
| CPXV | HumLue09/1 | Human | 2009 | KC813494 | Lübeck, Germany | [28] | |
| CPXV | GER_1990_2 | Human | 1990 | HQ420896 | Bonn, Germany | [27] | |
| CPXV | France-01 NANCY | Human | 2001 | HQ420894 | Nancy, France | [27] | |
| CPXV | NOR 1995 feline | Cat | 1994 | KY549151 | Bergen, Norway | E4 | This paper |
| CPXV | NOR 1994_MAN | Human | 1994 | HQ420899 | Bergen, Norway | [27] | |
| CPXV | UK 2000 K 1639 | Cat | 2000 | KY549148 | Bristol, UK | E5 | This paper |
| CPXV | UK 2000 K 2739 | Cat | 2000 | KY549149 | Bristol, UK | This paper | |
| CPXV | UK pox 5/wv1 | Cheetah | 1972 | KY549144 | London, UK | This paper | |
| CPXV | UK 2000 K 4207 | Cat | 2000 | KY549150 | Bristol, UK | This paper | |
| CPXV | UK_Brighton Red | Human | 1937 | AF482758 | Brighton, UK | [48] | |
| CPXV | UK 2000 K 2984 | Cat | 2000 | HQ420900 | Bristol, UK | [27] | |
| CPXV | UK 2000 K 428 | Cat | 2000 | KY549145 | Bristol, UK | This paper | |
| CPXV | UK 2000 K 779 | Cat | 2000 | KY549146 | Bristol, UK | E6 | This paper |
| CPXV | UK 2000 K 780 | Cat | 2000 | KY549147 | Bristol, UK | This paper | |
| CPXV | GER 1980-EP4 | Elephant | 1980 | HQ420895 | Hameln, Germany | [27] | |
| CPXV | HumPad07/1 | Human | 2007 | KC813496 | Paderborn, Germany | [28] | |
| CPXV | GER 2002 MKY | Marmoset | 2002 | HQ420898 | Gӧttingen, Germany | E7 | [27] |
| CPXV | BeaBer04/1 | Beaver | 2004 | KC813491 | Berlin, Germany | E8 | [28] |
| CPXV | CatBer07/1 | Cat | 2007 | KC813502 | Berlin, Germany | [28] | |
| CPXV | HumBer07/1 | Human | 2007 | KC813509 | Berlin, Germany | [28] | |
| CPXV | HumMag07/1 | Human | 2007 | KC813495 | Magdeburg, Germany | [28] | |
| CPXV | CatPot07/1 | Cat | 2007 | KC813506 | Potsdam, Germany | [28] | |
| CPXV | Amadeus_2015 | Horse | 2015 | LN879483 | Germany | [31] | |
| CPXV | EleGri07/1 Δ | Elephant | 2007 | KC813507 | Grimmen, Germany | E9 | [28] |
| CPXV | HumGri07/1 Δ | Human | 2007 | KC813511 | Grimmen, Germany | [28] | |
| CPXV | RatHei09/1 ‡ | Rat | 2009 | KC813504 | Heidelberg, Germany | E10 | [28] |
| CPXV | JagKre08/1 ‡ | Jaguarundi | 2008 | KC813497 | Krefeld, Germany | [28] | |
| CPXV | JagKre08/2 ‡ | Jaguarundi | 2008 | KC813498 | Krefeld, Germany | [28] | |
| CPXV | MonKre08/4 ‡ | Mongoose | 2008 | KC813500 | Krefeld, Germany | [28] | |
| CPXV | HumLan08/1 ‡ | Human | 2008 | KC813492 | Landau, Germany | [28] | |
| CMLV | M-96 | Camel | 1996 | NC003391 | Kazakhstan | CMLV | [49] |
| CMLV | CMS | Camel | 1970 | AY009089 | Iran | [50] | |
| ECTV | Naval | Mouse | 1996 | KJ563295 | USA | ECTV | [51] |
| ECTV | Moscow | Mouse | 1947 | NC004105 | Moscow, Russia | [52] | |
| MPXV | Liberia_1970 | Human | 1970 | DQ011156 | Liberia | MPXV | [53] |
| MPXV | Zaire-96 | Human | 1996 | NC003310 | Zaire/DRC | [54] | |
| TATV | Dahomey 1968 | Gerbil | 1968 | NC008291 | Dahomey, Benin | TATV | [47] |
| VACV | Rabbitpox Utrecht | Rabbit | 1941 | AY484669 | Utrecht, The Netherlands | VACV | [55] |
| VACV | Horsepox_MNR-76 | Horse | 1976 | DQ792504 | Mongolia | [56] | |
| VARV | Bangladesh1974 | Human | 1976 | DQ441420 | Bangladesh | VARV | [47] |
| VARV | Brazil1966 | Human | 1966 | DQ441419 | Brazil | [47] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauldin, M.R.; Antwerpen, M.; Emerson, G.L.; Li, Y.; Zoeller, G.; Carroll, D.S.; Meyer, H. Cowpox virus: What’s in a Name? Viruses 2017, 9, 101. https://doi.org/10.3390/v9050101
Mauldin MR, Antwerpen M, Emerson GL, Li Y, Zoeller G, Carroll DS, Meyer H. Cowpox virus: What’s in a Name? Viruses. 2017; 9(5):101. https://doi.org/10.3390/v9050101
Chicago/Turabian StyleMauldin, Matthew R., Markus Antwerpen, Ginny L. Emerson, Yu Li, Gudrun Zoeller, Darin S. Carroll, and Hermann Meyer. 2017. "Cowpox virus: What’s in a Name?" Viruses 9, no. 5: 101. https://doi.org/10.3390/v9050101
APA StyleMauldin, M. R., Antwerpen, M., Emerson, G. L., Li, Y., Zoeller, G., Carroll, D. S., & Meyer, H. (2017). Cowpox virus: What’s in a Name? Viruses, 9(5), 101. https://doi.org/10.3390/v9050101