
Targeting Pattern Recognition Receptors (PRR) for Vaccine Adjuvantation: From Synthetic PRR Agonists to the Potential of Defective Interfering Particles of Viruses

 ,
, 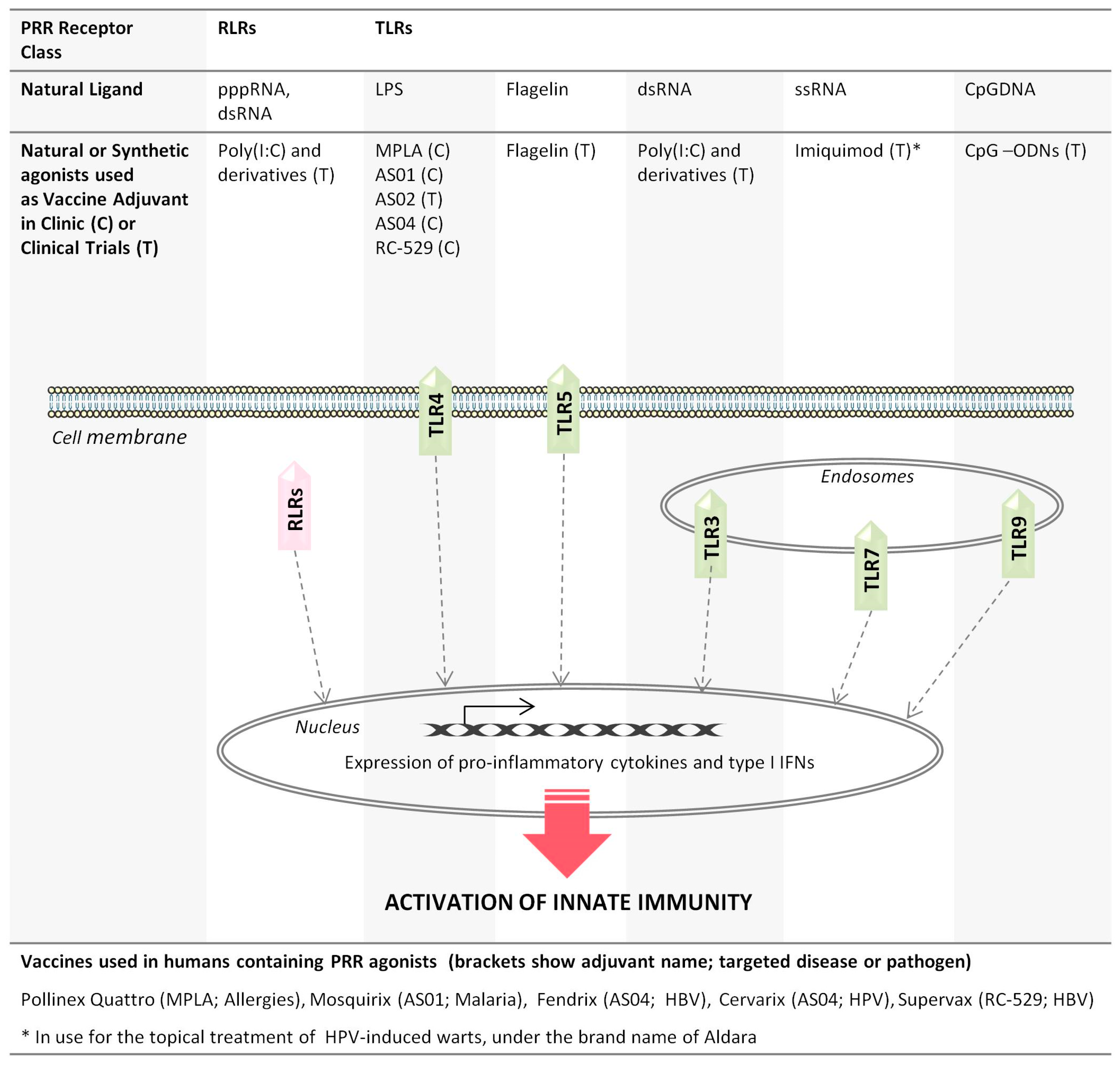{kind=link}
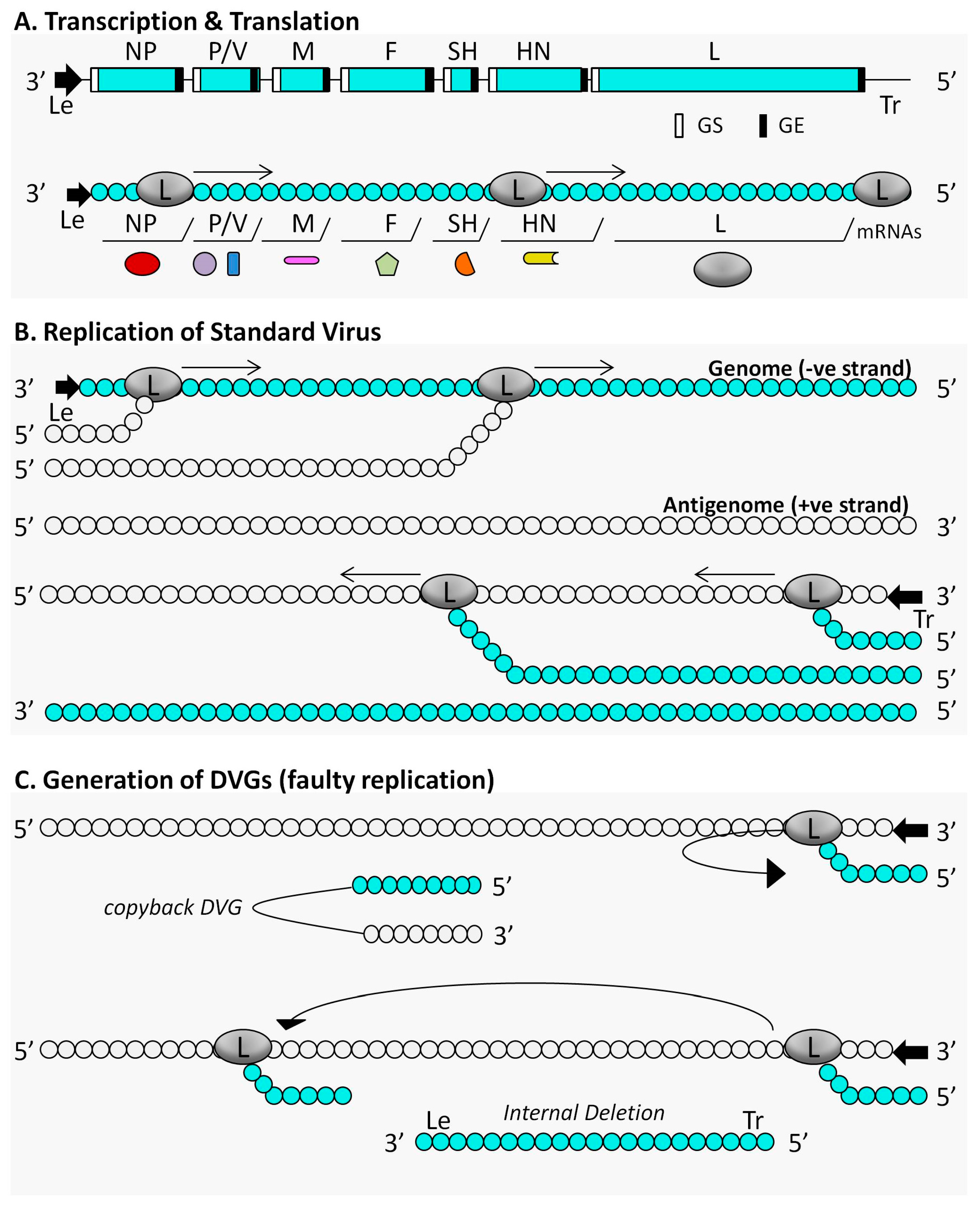{kind=link}
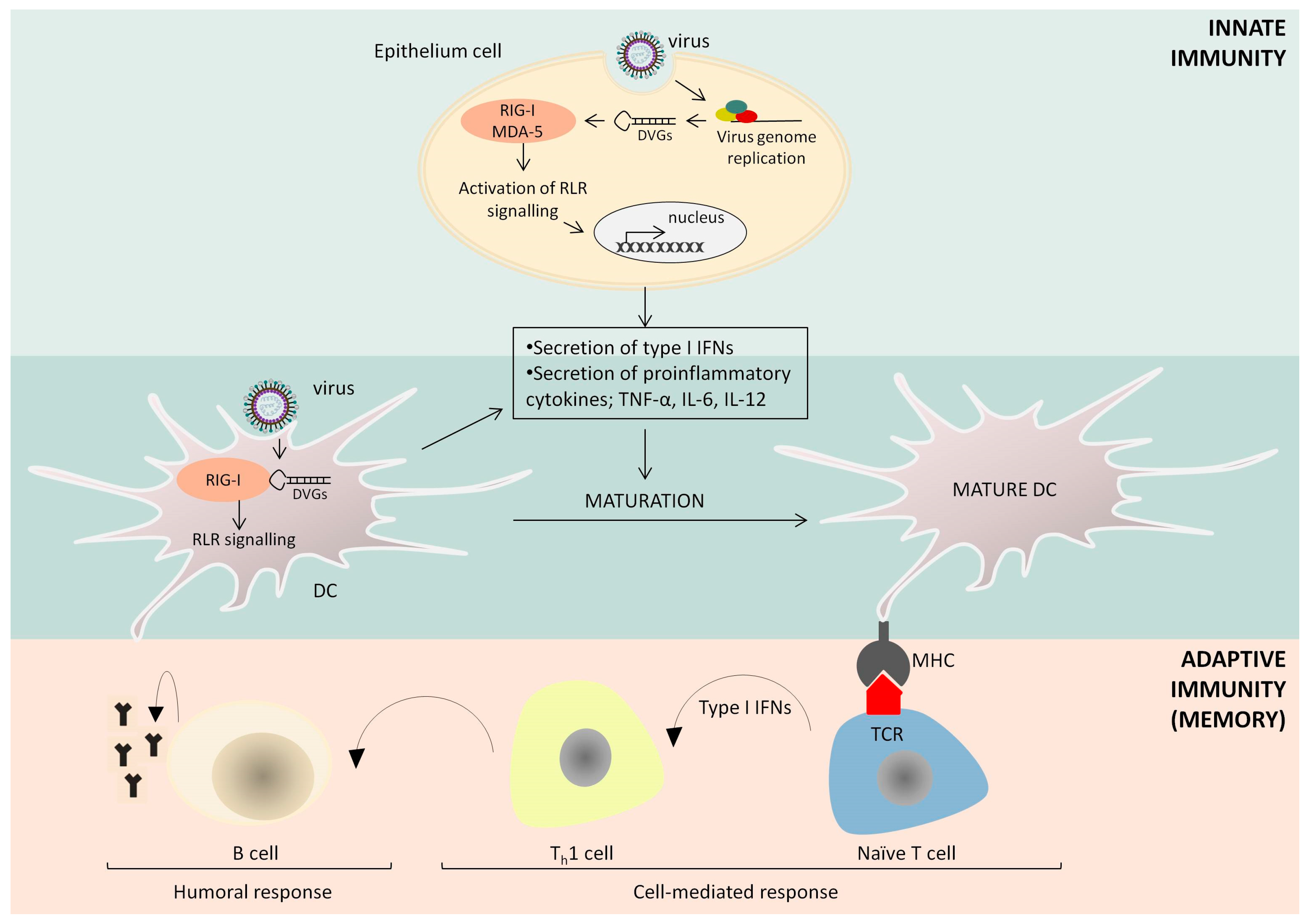{kind=link}
Abstract
:1. Making Better Vaccines; Vaccine Adjuvants
2. Pattern Recognition Receptor Agonists: A Diverse Class of Vaccine Adjuvants
3. The Immunostimulatory Activity of Defective Interfering Particles of Negative-Sense RNA Viruses
4. Further Applications of Defective Interfering Particles in Vaccine Adjuvantation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ribeiro, C.M.; Schijns, V.E. Immunology of vaccine adjuvants. Methods Mol. Biol. 2010, 626, 1–14. [Google Scholar] [PubMed]
- Eibl, M.M.; Wolf, H.M. Vaccination in patients with primary immune deficiency, secondary immune deficiency and autoimmunity with immune regulatory abnormalities. Immunotherapy 2015, 7, 1273–1292. [Google Scholar] [CrossRef] [PubMed]
- Glenny, A.T.; Barr, M. The precipitation of diphtheria toxoid by potash alum. J. Pathol. Bacteriol. 1931, 34, 131–138. [Google Scholar] [CrossRef]
- Park, W.H.; Schroder, M.C. Diphtheria Toxin-Antitoxin and Toxoid: A Comparison. Am. J. Public Health Nat. Health 1932, 22, 7–16. [Google Scholar] [CrossRef]
- Stephenson, I.; Nicholson, K.G.; Colegate, A.; Podda, A.; Wood, J.; Ypma, E.; Zambon, M. Boosting immunity to influenza H5N1 with MF59-adjuvanted H5N3 A/Duck/Singapore/97 vaccine in a primed human population. Vaccine 2003, 21, 1687–1693. [Google Scholar] [CrossRef]
- Tong, N.K.C.; Beran, J.; Kee, S.A.; Miguel, J.L.; Sánchez, C.; Bayas, J.M.; Vilella, A.; de Juanes, J.R.; Arrazola, P.; Calbo-Torrecillas, F.; et al. Immunogenicity and safety of an adjuvanted hepatitis B vaccine in pre-hemodialysis and hemodialysis patients. Kidney Int. 2005, 68, 2298–2303. [Google Scholar] [CrossRef] [PubMed]
- Baz, M.; Luke, C.J.; Cheng, X.; Jin, H.; Subbarao, K. H5N1 vaccines in humans. Virus Res. 2013, 178, 78–98. [Google Scholar] [CrossRef] [PubMed]
- Black, R.E.; Cousens, S.; Johnson, H.L.; Lawn, J.E.; Rudan, I.; Bassani, D.G.; Jha, P.; Campbell, H.; Walker, C.F.; Cibulskis, R.; et al. Global, regional, and national causes of child mortality in 2008: A systematic analysis. Lancet 2010, 375, 1969–1987. [Google Scholar] [CrossRef]
- World Health Organization. The Top 10 Causes of Death. Available online: http://www.who.int/mediacentre/factsheets/fs310/en/ (accessed on 17 April 2017).
- Di Pasquale, A.; Preiss, S.; Tavares Da Silva, F.; Garcon, N. Vaccine Adjuvants: From 1920 to 2015 and Beyond. Vaccines 2015, 3, 320–343. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.P.; Farrar, D.J. Regulation of effector and memory T-cell functions by type I interferon. Immunology 2011, 132, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Leo, O. Key concepts in immunology. Vaccine 2010, 28, C2–C13. [Google Scholar] [CrossRef] [PubMed]
- Awate, S.; Babiuk, L.A.; Mutwiri, G. Mechanisms of action of adjuvants. Front. Immunol. 2013, 4, 114. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, E.J.; Tan, A.C.L.; Jackson, D.C. TLR agonists as modulators of the innate immune response and their potential as agents against infectious disease. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, N.J.; Easton, A.J. Defective interfering influenza virus RNAs: Time to reevaluate their clinical potential as broad-spectrum antivirals? J. Virol. 2014, 88, 5217–5227. [Google Scholar] [CrossRef] [PubMed]
- Gutjahr, A.; Tiraby, G.; Perouzel, E.; Verrier, B.; Paul, S. Triggering Intracellular Receptors for Vaccine Adjuvantation. Trends Immunol. 2016, 37, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Apostolico, J.D.S.; Lunardelli, V.A.S.; Coirada, F.C.; Boscardin, S.B.; Rosa, D.S. Adjuvants: Classification, Modus Operandi, and Licensing. J. Immunol. Res. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hedayat, M.; Netea, M.G.; Rezaei, N. Targeting of Toll-like receptors: A decade of progress in combating infectious diseases. Lancet Infect. Dis. 2011, 11, 702–712. [Google Scholar] [CrossRef]
- Dowling, J.K.; Mansell, A. Toll-like receptors: The swiss army knife of immunity and vaccine development. Clin. Transl. Immunol. 2016, 5, e85. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol. Med. 2011, 3, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G.; Sher, A. Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 2007, 7, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, E.; Yang, D.; Lu, M. Contribution of Toll-like receptors to the control of hepatitis B virus infection by initiating antiviral innate responses and promoting specific adaptive immune responses. Cell. Mol. Immunol. 2015, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J.; Harper, D.M. Human papillomavirus types 16 and 18 vaccine (recombinant, AS04 adjuvanted, adsorbed) [Cervarix]. Drugs 2008, 68, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Garçon, N.; Chomez, P.; Van Mechelen, M. GlaxoSmithKline Adjuvant Systems in vaccines: Concepts, achievements and perspectives. Expert Rev. Vaccines 2007, 6, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Casella, C.R.; Mitchell, T.C. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell. Mol. Life Sci. CMLS 2008, 65, 3231–3240. [Google Scholar] [CrossRef] [PubMed]
- Didierlaurent, A.M.; Morel, S.; Lockman, L.; Giannini, S.L.; Bisteau, M.; Carlsen, H.; Kielland, A.; Vosters, O.; Vanderheyde, N.; Schiavetti, F.; et al. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. 2009, 183, 6186–6197. [Google Scholar] [CrossRef] [PubMed]
- Garçon, N. Preclinical development of AS04. Methods Mol. Biol. 2010, 626, 15–27. [Google Scholar] [PubMed]
- First Malaria Vaccine Receives Positive Scientific Opinion from EMA. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/07/news_detail_002376.jsp&mid=WC0b01ac058004d5c1 (accessed on 27 April 2017).
- Kester, K.E.; Cummings, J.F.; Ofori-Anyinam, O.; Ockenhouse, C.F.; Krzych, U.; Moris, P.; Schwenk, R.; Nielsen, R.A.; Debebe, Z.; Pinelis, E.; et al. Randomized, Double-Blind, Phase 2a Trial of Falciparum Malaria Vaccines RTS,S/AS01B and RTS,S/AS02A in Malaria-Naive Adults: Safety, Efficacy, and Immunologic Associates of Protection. J. Infect. Dis. 2009, 200, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Leroux-Roels, G.; Leroux-Roels, I.; Clement, F.; Ofori-Anyinam, O.; Lievens, M.; Jongert, E.; Moris, P.; Ballou, W.R.; Cohen, J. Evaluation of the immune response to RTS,S/AS01 and RTS,S/AS02 adjuvanted vaccines: Randomized, double-blind study in malaria-naive adults. Hum. Vaccines Immunother. 2014, 10, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Rosewich, M.; Lee, D.; Zielen, S. Pollinex Quattro: An innovative four injections immunotherapy In allergic rhinitis. Hum. Vaccines Immunother. 2013, 9, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, J.R.; Cluff, C.W.; Evans, J.T.; Lacy, M.J.; Stephens, J.R.; Brookshire, V.G.; Wang, R.; Ward, J.R.; Yorgensen, Y.M.; Persing, D.H.; et al. Immunostimulatory activity of aminoalkyl glucosaminide 4-phosphates (AGPs): Induction of protective innate immune responses by RC-524 and RC-529. J. Endotoxin Res. 2002, 8, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Dupont, J.; Altclas, J.; Lepetic, A.; Lombardo, M.; Vázquez, V.; Salgueira, C.; Seigelchifer, M.; Arndtz, N.; Antunez, E.; von Eschen, K.; et al. A controlled clinical trial comparing the safety and immunogenicity of a new adjuvanted hepatitis B vaccine with a standard hepatitis B vaccine. Vaccine 2006, 24, 7167–7174. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Dinehart, S.; Whiting, D.; Lee, P.K.; Tawfik, N.; Jorizzo, J.; Lee, J.H.; Fox, T.L. Imiquimod 5% cream for the treatment of actinic keratosis: Results from two phase III, randomized, double-blind, parallel group, vehicle-controlled trials. J. Am. Acad. Dermatol. 2004, 50, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.F.N.; Zhang, A.J.; To, K.K.W.; Chan, J.F.W.; Li, C.; Zhu, H.S.; Li, P.; Chan, T.C.; Cheng, V.C.C.; Chan, K.H.; et al. Immunogenicity of Intradermal Trivalent Influenza Vaccine With Topical Imiquimod: A Double Blind Randomized Controlled Trial. Clin. Infect. Dis. 2014, 59, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Therapeutic potential of Toll-like receptor 9 activation. Nat. Rev. Drug Discov. 2006, 5, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.L.; Davis, H.L.; Morrris, M.L.; Efler, S.M.; Adhami, M.A.; Krieg, A.M.; Cameron, D.W.; Heatcote, J. CPG 7909, an Immunostimulatory TLR9 Agonist Oligodeoxynucleotide, as Adjuvant to Engerix-B HBV Vaccine in Healthy Adults: A Double-Blind Phase I/II Study. J. Clin. Immunol. 2004, 24, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-Z.; Ma, Y.; Xu, W.-H.; Wang, S.; Sun, Z.-W. Combinations of various CpG motifs cloned into plasmid backbone modulate and enhance protective immunity of viral replicon DNA anthrax vaccines. Med. Microbiol. Immunol. 2015, 204, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Turley, C.B.; Rupp, R.E.; Johnson, C.; Taylor, D.N.; Wolfson, J.; Tussey, L.; Kavita, U.; Stanberry, L.; Shaw, A. Safety and immunogenicity of a recombinant M2e–flagellin influenza vaccine (STF2.4xM2e) in healthy adults. Vaccine 2011, 29, 5145–5152. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.N.; Treanor, J.J.; Sheldon, E.A.; Johnson, C.; Umlauf, S.; Song, L.; Kavita, U.; Liu, G.; Tussey, L.; Ozer, K.; et al. Development of VAX128, a recombinant hemagglutinin (HA) influenza-flagellin fusion vaccine with improved safety and immune response. Vaccine 2012, 30, 5761–5769. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, C.; Medzhitov, R.; Flavell, R. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Diebold, S.S.; Chen, M.; Näslund, T.I.; Nolte, M.A.; Alexopoulou, L.; Azuma, Y.-T.; Flavell, R.A.; Liljeström, P.; Reis e Sousa, C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, J.J.; Wolff, S.M.; Levy, H.B. Inhibition of biologic activity of poly I: Poly C by human plasma. Proc. Soc. Exp. Biol. Med. 1970, 133, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.B.; Riley, F.L.; Lvovsky, E.; Stephen, E.E. Interferon induction in primates by stabilized polyriboinosinic acid-polyribocytidylic acid: Effect of component size. Infect. Immun. 1981, 34, 416–421. [Google Scholar] [PubMed]
- Longhi, M.P.; Trumpfheller, C.; Idoyaga, J.; Caskey, M.; Matos, I.; Kluger, C.; Salazar, A.M.; Colonna, M.; Steinman, R.M. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 2009, 206, 1589–1602. [Google Scholar] [CrossRef] [PubMed]
- Stahl-Hennig, C.; Eisenblatter, M.; Jasny, E.; Rzehak, T.; Tenner-Racz, K.; Trumpfheller, C.; Salazar, A.M.; Uberla, K.; Nieto, K.; Kleinschmidt, J.; et al. Synthetic double-stranded RNAs are adjuvants for the induction of T helper 1 and humoral immune responses to human papillomavirus in rhesus macaques. PLoS Pathog. 2009, 5, e1000373. [Google Scholar] [CrossRef] [PubMed]
- Trumpfheller, C.; Caskey, M.; Nchinda, G.; Longhi, M.P.; Mizenina, O.; Huang, Y.; Schlesinger, S.J.; Colonna, M.; Steinman, R.M. The microbial mimic poly IC induces durable and protective CD4+ T cell immunity together with a dendritic cell targeted vaccine. Proc. Nat. Acad. Sci. USA 2008, 105, 2574–2579. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F. Failure at the effector phase: Immune barriers at the level of the melanoma tumor microenvironment. Clin. Cancer Res. 2007, 13, 5256–5261. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.-R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Ammi, R.; De Waele, J.; Willemen, Y.; Van Brussel, I.; Schrijvers, D.M.; Lion, E.; Smits, E.L. Poly(I:C) as cancer vaccine adjuvant: Knocking on the door of medical breakthroughs. Pharmacol. Ther. 2015, 146, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Butowski, N.; Chang, S.M.; Junck, L.; DeAngelis, L.M.; Abrey, L.; Fink, K.; Cloughesy, T.; Lamborn, K.R.; Salazar, A.M.; Prados, M.D. A phase II clinical trial of poly-ICLC with radiation for adult patients with newly diagnosed supratentorial glioblastoma: A North American Brain Tumor Consortium (NABTC01-05). J. Neuro-Oncol. 2009, 91, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K.; et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with α-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patie. J. Clin. Oncol. 2011, 29, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, S.; Britten, C.D.; Chin, S.; Garrett-Mayer, E.; Cloud, C.A.; Li, M.; Scurti, G.; Salem, M.L.; Nelson, M.H.; Thomas, M.B.; Paulos, C.M.; et al. Vaccination with poly(IC:LC) and peptide-pulsed autologous dendritic cells in patients with pancreatic cancer. J. Hematol. Oncol. 2017, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Caskey, M.; Lefebvre, F.; Filali-Mouhim, A.; Cameron, M.J.; Goulet, J.P.; Haddad, E.K.; Breton, G.; Trumpfheller, C.; Pollak, S.; Shimeliovich, I.; et al. Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J. Exp. Med. 2011, 208, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, R.; Jasny, E.; Kowalczyk, A.; Lutz, J.; Probst, J.; Baumhof, P.; Scheel, B.; Voss, S.; Kallen, K.-J.; Fotin-Mleczek, M. A novel RNA-based adjuvant combines strong immunostimulatory capacities with a favorable safety profile. Int. J. Cancer 2015, 137, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Circelli, L.; Petrizzo, A.; Tagliamonte, M.; Heidenreich, R.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Immunological effects of a novel RNA-based adjuvant in liver cancer patients. Cancer Immunol. Immunother. 2016, 66, 1–10. [Google Scholar] [CrossRef]
- Huang, A.S.; Baltimore, D. Defective Viral Particles and Viral Disease Processes. Nature 1970, 226, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, R.A.; Keene, J.D.; Schubert, M. The origins of defective interfering particles of the negative-strand RNA viruses. Cell 1981, 26, 145–154. [Google Scholar] [CrossRef]
- Roux, L.; Simon, A.E.; Holland, J.J. Effects of defective interfering viruses on virus replication and pathogenesis in vitro and in vivo. Adv. Virus Res. 1991, 40, 181–211. [Google Scholar] [PubMed]
- Marriott, A.C.; Dimmock, N.J. Defective interfering viruses and their potential as antiviral agents. Rev. Med. Virol. 2010, 20, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Von Magnus, P. Studies on Interference in Experimental Influenza: Purification and Centrifugation Experiments. Ark. Kemi Mineral. Geol. 1947, 24b, 1–6. [Google Scholar]
- Pathak, K.B.; Nagy, P.D. Defective Interfering RNAs: Foes of Viruses and Friends of Virologists. Viruses 2009, 1, 895–919. [Google Scholar] [CrossRef] [PubMed]
- Nuesch, J.P.F.; De Chastonay, J.; Siegl, G. Detection of Defective Genomes in Hepatitis A Virus Particles Present in Clinical Specimens. J. Gen. Virol. 1989, 70, 3475–3480. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.T.; Lin, M.H.; Chen, D.S.; Shih, C. A defective interference-like phenomenon of human hepatitis B virus in chronic carriers. J. Virol. 1998, 72, 578–584. [Google Scholar] [PubMed]
- Yeh, C.T.; Chu, C.M.; Lu, S.C.; Liaw, Y.F. Molecular cloning of a defective hepatitis C virus genome from the ascitic fluid of a patient with hepatocellular carcinoma. J. Gen. Virol. 1997, 78, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lott, W.B.; Lowry, K.; Jones, A.; Thu, H.M.; Aaskov, J. Defective interfering viral particles in acute dengue infections. PLoS ONE 2011, 6, e19447. [Google Scholar] [CrossRef] [PubMed]
- Saira, K.; Lin, X.; DePasse, J.V.; Halpin, R.; Twaddle, A.; Stockwell, T.; Angus, B.; Cozzi-Lepri, A.; Delfino, M.; Dugan, V.; et al. Sequence Analysis of In Vivo Defective Interfering-Like RNA of Influenza A H1N1 Pandemic Virus. J. Virol. 2013, 87, 8064–8074. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jain, D.; Koziol-White, C.J.; Genoyer, E.; Gilbert, M.; Tapia, K.; Panettieri, R.A., Jr.; Hodinka, R.L.; Lopez, C.B. Immunostimulatory Defective Viral Genomes from Respiratory Syncytial Virus Promote a Strong Innate Antiviral Response during Infection in Mice and Humans. PLoS Pathog. 2015, 11, e1005122. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, N.J. The Biological Significance of Defective Interfering Viruses. Ann. Rev. Microbiol. 1991, 1, 165–176. [Google Scholar] [CrossRef]
- Hsu, C.H.; Re, G.G.; Gupta, K.C.; Portner, A.; Kingsbury, D.W. Expression of sendai virus defective-interfering genomes with internal deletions. Virology 1985, 146, 38–49. [Google Scholar] [CrossRef]
- Re, G.G.; Morgan, E.M.; Kingsbury, D.W. Nucleotide sequences responsible for generation of internally deleted Sendai virus defective interfering genomes. Virology 1985, 146, 27–37. [Google Scholar] [CrossRef]
- Yount, J.S.; Gitlin, L.; Moran, T.M.; López, C.B. MDA5 participates in the detection of paramyxovirus infection and is essential for the early activation of dendritic cells in response to Sendai Virus defective interfering particles. J. Immunol. 2008, 180, 4910–4918. [Google Scholar] [CrossRef] [PubMed]
- Tapia, K.; Kim, W. k.; Sun, Y.; Mercado-Lopez, X.; Dunay, E.; Wise, M.; Adu, M.; Lopez, C.B. Defective Viral Genomes Arising In Vivo Provide Critical Danger Signals for the Triggering of Lung Antiviral Immunity. PLoS Pathog. 2013, 9, e1003703. [Google Scholar] [CrossRef] [PubMed]
- Yount, J.S.; Kraus, T.A.; Horvath, C.M.; Moran, T.M.; Lopez, C.B. A Novel Role for Viral-Defective Interfering Particles in Enhancing Dendritic Cell Maturation. J. Immunol. 2006, 177, 4503–4513. [Google Scholar] [CrossRef] [PubMed]
- Killip, M.J.; Young, D.F.; Gatherer, D.; Ross, C.S.; Short, J.A.L.; Davison, A.J.; Goodbourn, S.; Randall, R.E. Deep sequencing analysis of defective genomes of parainfluenza virus 5 and their role in interferon induction. J. Virol. 2013, 87, 4798–4807. [Google Scholar] [CrossRef] [PubMed]
- Mercado-Lopez, X.; Cotter, C.R.; Kim, W.K.; Sun, Y.; Munoz, L.; Tapia, K.; Lopez, C.B. Highly immunostimulatory RNA derived from a Sendai virus defective viral genome. Vaccine 2013, 31, 5713–5721. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Gil, L.; Goff, P.H.; Hai, R.; García-Sastre, A.; Shaw, M.L.; Palese, P. A Sendai virus-derived RNA agonist of RIG-I as a virus vaccine adjuvant. J. Virol. 2013, 87, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Mercado-Lopez, X.; Grier, J.T.; Kim, W.K.; Chun, L.F.; Irvine, E.B.; Duany, Y.D.T.; Kell, A.; Hur, S.; Gale, M.; et al. Identification of a natural viral RNA motif that optimizes sensing of viral RNA by RIG-I. mBio 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Sachidanandam, R.; Garcia-Sastre, A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Nat. Acad. Sci. USA 2010, 107, 16303–16308. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Strahle, L.; Garcin, D.; Kolakofsky, D. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 2006, 351, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Short, J.A.; Young, D.F.; Killip, M.J.; Schneider, M.; Goodbourn, S.; Randall, R.E. Heterocellular induction of interferon by negative-sense RNA viruses. Virology 2010, 407, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Killip, M.J.; Young, D.F.; Precious, B.L.; Goodbourn, S.; Randall, R.E. Activation of the beta interferon promoter by paramyxoviruses in the absence of virus protein synthesis. J. Gen. Virol. 2012, 93, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Killip, M.J.; Young, D.F.; Ross, C.S.; Chen, S.; Goodbourn, S.; Randall, R.E. Failure to activate the IFN-beta promoter by a paramyxovirus lacking an interferon antagonist. Virology 2011, 415, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, N.J.; Easton, A.J. Cloned Defective Interfering Influenza RNA and a Possible Pan-Specific Treatment of Respiratory Virus Diseases. Viruses 2015, 7, 3768–3788. [Google Scholar] [CrossRef] [PubMed]
- Easton, A.J.; Scott, P.D.; Edworthy, N.L.; Meng, B.; Marriott, A.C.; Dimmock, N.J. A novel broad-spectrum treatment for respiratory virus infections: Influenza-based defective interfering virus provides protection against pneumovirus infection in vivo. Vaccine 2011, 29, 2777–2784. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.D.; Meng, B.; Marriott, A.C.; Easton, A.J.; Dimmock, N.J. Defective interfering influenza A virus protects in vivo against disease caused by a heterologous influenza B virus. J. Gen. Virol. 2011, 92, 2122–2132. [Google Scholar] [CrossRef] [PubMed]
- Clavell, L.A.; Bratt, M.A. Relationship between the ribonucleic acid synthesizing capacity of ultraviolet-irradiated Newcastle disease virus and its ability to induce interferon. J. Virol. 1971, 8, 500–508. [Google Scholar] [PubMed]
- Beljanski, V.; Chiang, C.; Kirchenbaum, G.A.; Olagnier, D.; Bloom, C.E.; Wong, T.; Haddad, E.K.; Trautmann, L.; Ross, T.M.; Hiscott, J. Enhanced Influenza Virus-Like Particle Vaccination with a Structurally Optimized RIG-I Agonist as Adjuvant. J. Virol. 2015, 89, 10612–10624. [Google Scholar] [CrossRef] [PubMed]
- McLaren, L.C.; Holland, J.J. Defective interfering particles from poliovirus vaccine and vaccine reference strains. Virology 1974, 60, 579–583. [Google Scholar] [CrossRef]
- Shingai, M.; Ebihara, T.; Begum, N.A.; Kato, A.; Honma, T.; Matsumoto, K.; Saito, H.; Ogura, H.; Matsumoto, M.; Seya, T. Differential type I IFN-inducing abilities of wild-type versus vaccine strains of measles virus. J. Immunol. 2007, 179, 6123–6133. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.-H.; Kew, C.; Lui, P.-Y.; Chan, C.-P.; Satoh, T.; Akira, S.; Jin, D.-Y.; Kok, K.-H. PACT- and RIG-I-Dependent Activation of Type I Interferon Production by a Defective Interfering RNA Derived from Measles Virus Vaccine. J. Virol. 2016, 90, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Dudek, T.; Knipe, D.M. Replication-defective viruses as vaccines and vaccine vectors. Virology 2006, 344, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U. Mechanisms of RIG-I-like receptor activation and manipulation by viral pathogens. J. Virol. 2014, 88, 5213–5216. [Google Scholar] [CrossRef] [PubMed]
- McClure, R.; Massari, P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Front. Immunol. 2014, 5, 386. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, P.; Wu, M.X. Natural STING Agonist as an "Ideal" Adjuvant for Cutaneous Vaccination. J. Investig. Dermatol. 2016, 136, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- CDC. CDC’s Strategic Framework for Global Immunization, 2016–2020; CDC: Atlanta, GA, USA, 2016. [Google Scholar]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasou, A.; Sultanoglu, N.; Goodbourn, S.; Randall, R.E.; Kostrikis, L.G. Targeting Pattern Recognition Receptors (PRR) for Vaccine Adjuvantation: From Synthetic PRR Agonists to the Potential of Defective Interfering Particles of Viruses. Viruses 2017, 9, 186. https://doi.org/10.3390/v9070186
Vasou A, Sultanoglu N, Goodbourn S, Randall RE, Kostrikis LG. Targeting Pattern Recognition Receptors (PRR) for Vaccine Adjuvantation: From Synthetic PRR Agonists to the Potential of Defective Interfering Particles of Viruses. Viruses. 2017; 9(7):186. https://doi.org/10.3390/v9070186
Chicago/Turabian StyleVasou, Andri, Nazife Sultanoglu, Stephen Goodbourn, Richard E. Randall, and Leondios G. Kostrikis. 2017. "Targeting Pattern Recognition Receptors (PRR) for Vaccine Adjuvantation: From Synthetic PRR Agonists to the Potential of Defective Interfering Particles of Viruses" Viruses 9, no. 7: 186. https://doi.org/10.3390/v9070186
APA StyleVasou, A., Sultanoglu, N., Goodbourn, S., Randall, R. E., & Kostrikis, L. G. (2017). Targeting Pattern Recognition Receptors (PRR) for Vaccine Adjuvantation: From Synthetic PRR Agonists to the Potential of Defective Interfering Particles of Viruses. Viruses, 9(7), 186. https://doi.org/10.3390/v9070186