
The Telomeric Response to Viral Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
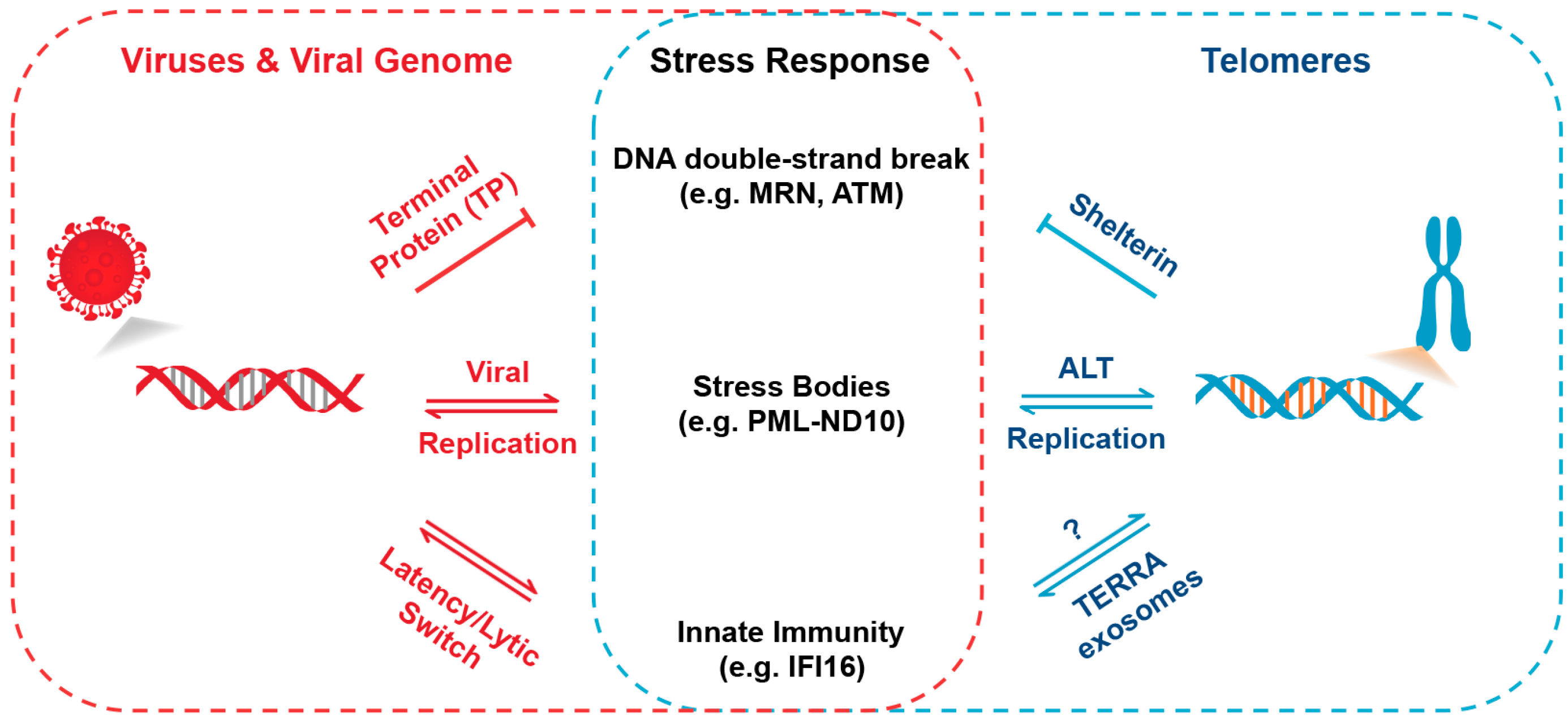
:1. Introduction
2. Telomeres as Functional Genetic Elements
3. Telomeric Chromatin Structure and Conformation
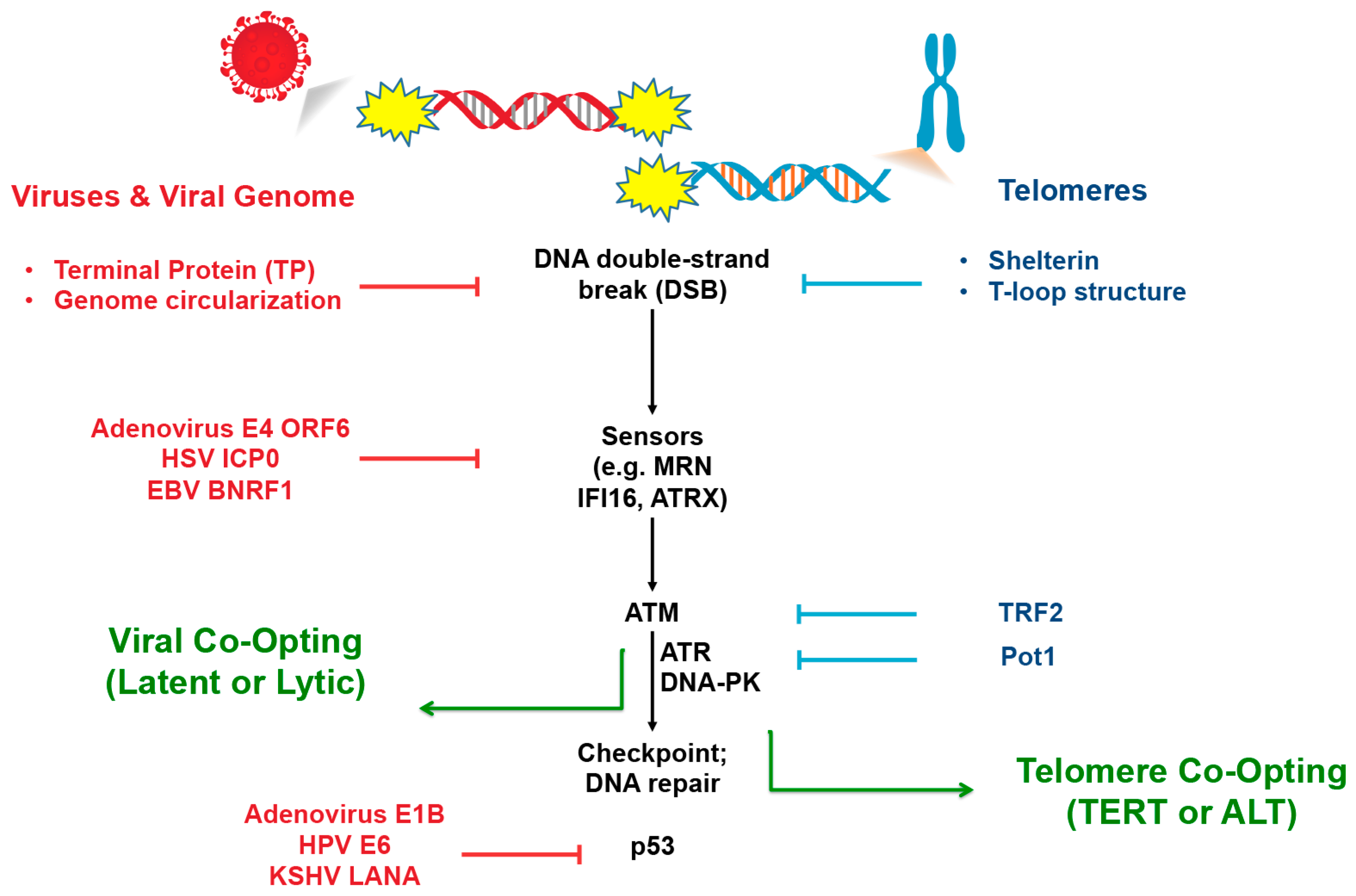
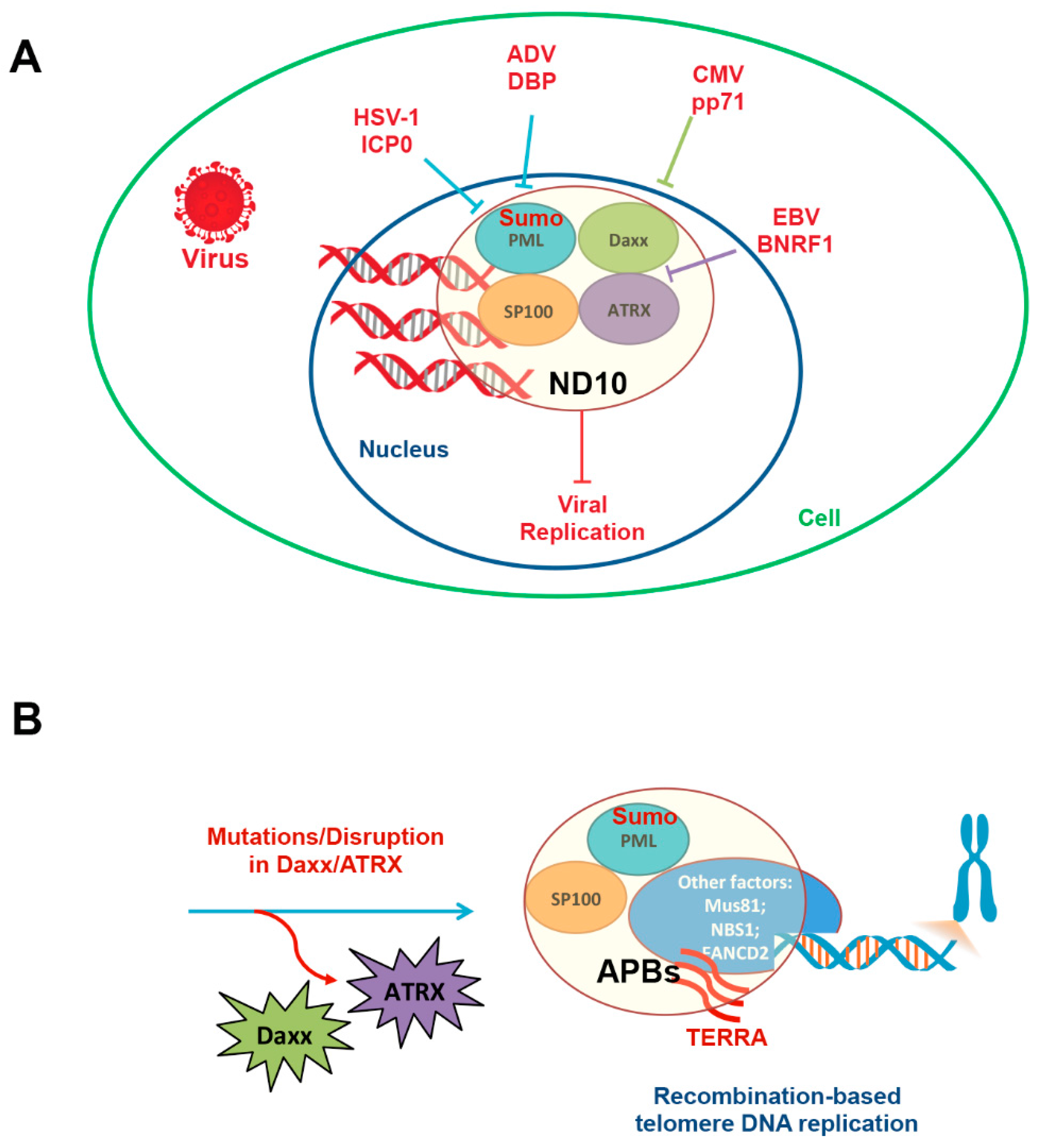
4. Viral Recapitulation of Telomere End-Protection
5. Virus Infection Can Alter Telomere Maintenance
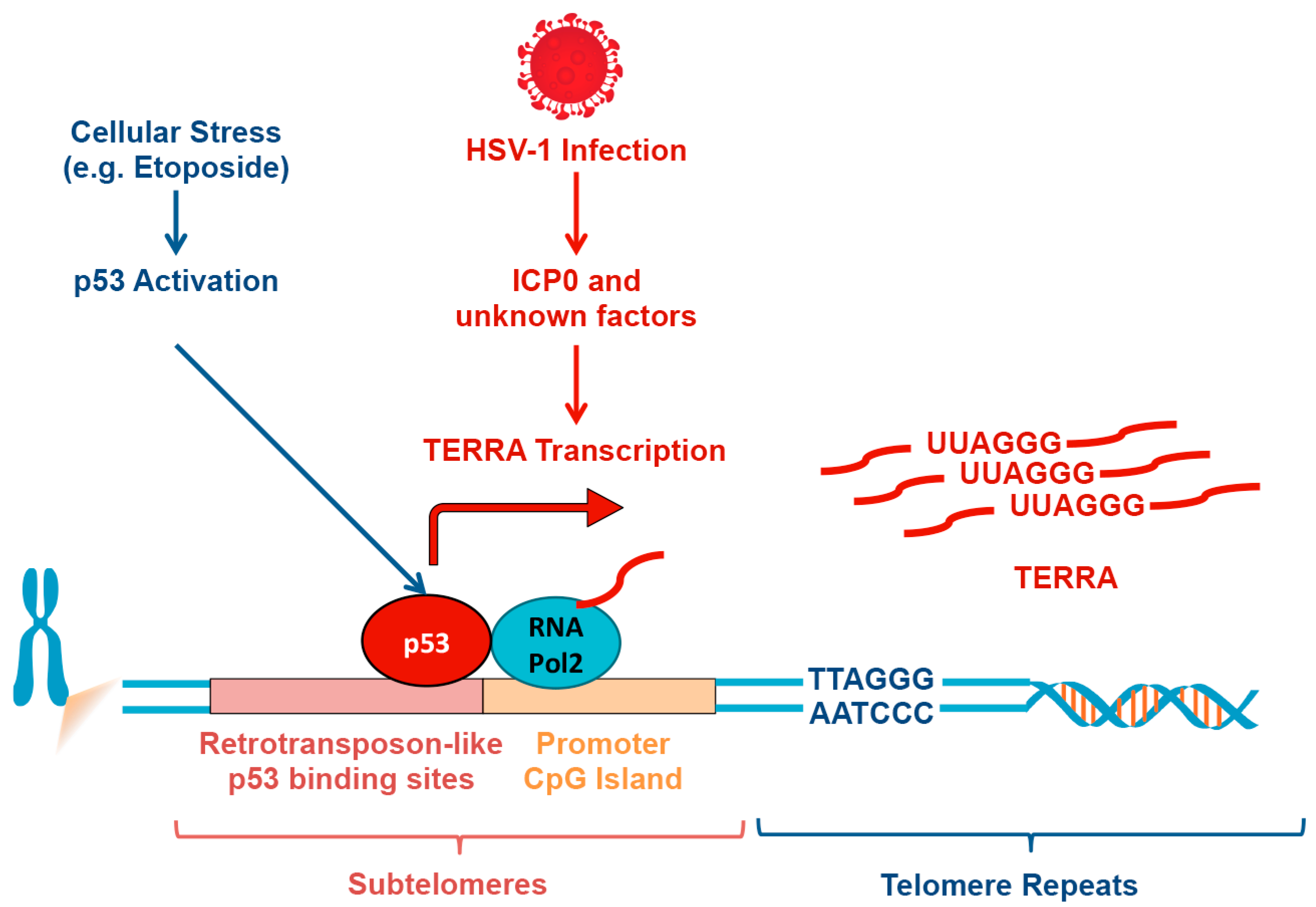
6. TERRA Induction by p53 Response Elements in Subtelomeres
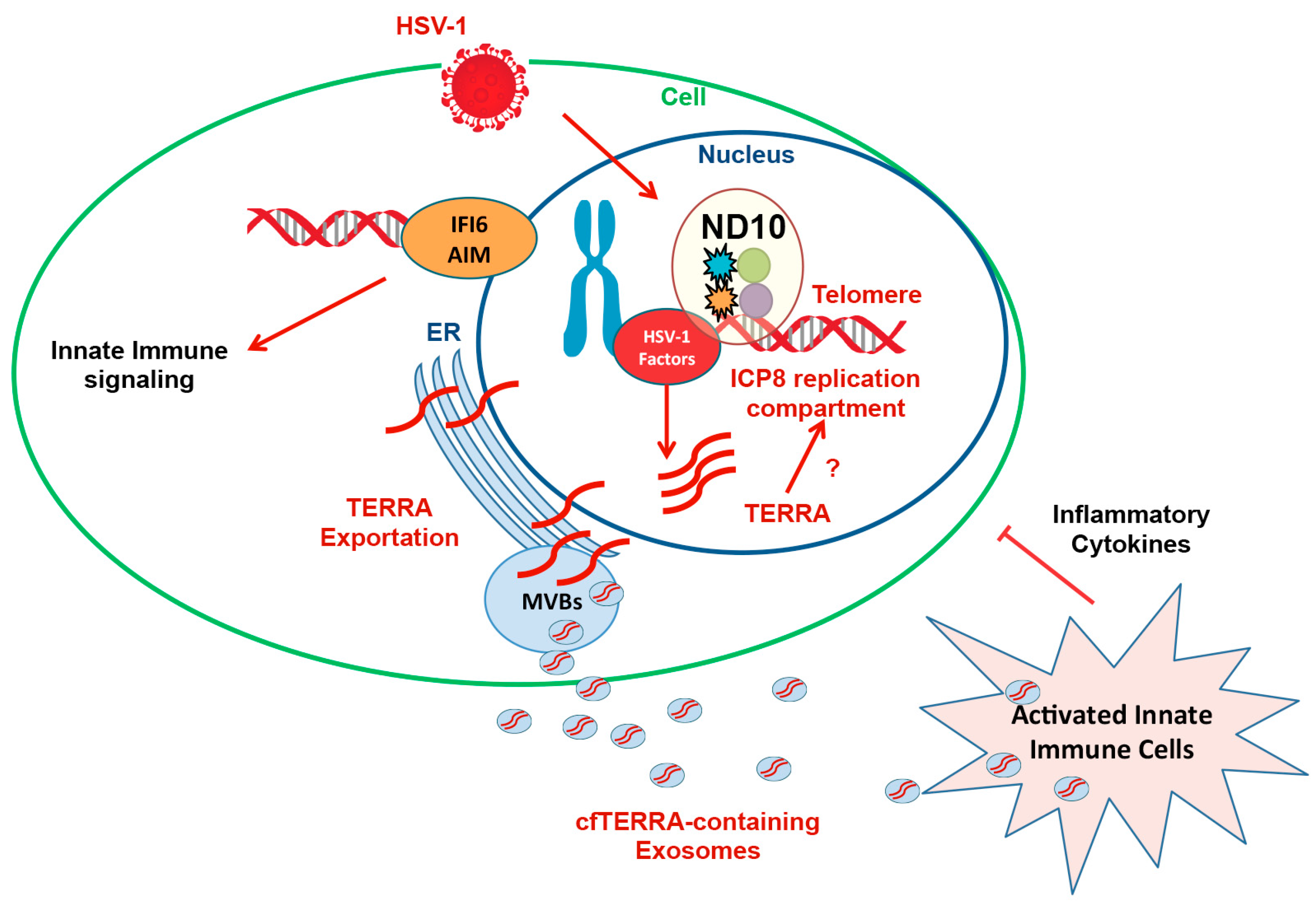
7. Antiviral Signaling Properties of TERRA
8. Conclusions
Conflicts of Interest
References
- Deng, Z.; Wang, Z.; Lieberman, P.M. Telomeres and viruses: Common themes of genome maintenance. Front. Oncol. 2012, 2, 201. [Google Scholar] [CrossRef] [PubMed]
- Boscolo-Rizzo, P.; Da Mosto, M.C.; Rampazzo, E.; Giunco, S.; Del Mistro, A.; Menegaldo, A.; Baboci, L.; Mantovani, M.; Tirelli, G.; De Rossi, A. Telomeres and telomerase in head and neck squamous cell carcinoma: From pathogenesis to clinical implications. Cancer Metast. Rev. 2016, 35, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Senkevich, T.G.; Katsafanas, G.; Weisberg, A.; Olano, L.R.; Moss, B. Identification of Vaccinia Virus Replisome and Transcriptome Proteins by iPOND Coupled with Mass Spectrometry. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. A loopy view of telomere evolution. Front. Genet. 2015, 6, 321. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomeres and telomerase: Their mechanisms of action and the effects of altering their functions. FEBS Lett. 2005, 579, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R. Beginning to understand the end of the chromosome. Cell 2004, 116, 273–279. [Google Scholar] [CrossRef]
- Feldser, D.M.; Hackett, J.A.; Greider, C.W. Telomere dysfunction and the initiation of genome instability. Nat. Rev. Cancer 2003, 3, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.A.; Upton, H.E.; Vogan, J.M.; Collins, K. Telomerase Mechanism of Telomere Synthesis. Annu. Rev. Biochem. 2017, 86, 439–460. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Villasante, A.; de Pablos, B.; Mendez-Lago, M.; Abad, J.P. Telomere maintenance in Drosophila: Rapid transposon evolution at chromosome ends. Cell Cycle 2008, 7, 2134–2138. [Google Scholar] [CrossRef] [PubMed]
- Cusanelli, E.; Chartrand, P. Telomeric repeat-containing RNA TERRA: A noncoding RNA connecting telomere biology to genome integrity. Front. Genet. 2015, 6, 143. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, C.M.; Lingner, J. Telomere functions grounding on TERRA firma. Trends Cell Biol. 2015, 25, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Luke, B.; Lingner, J. TERRA: Telomeric repeat-containing RNA. EMBO J. 2009, 28, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Diman, A.; Boros, J.; Poulain, F.; Rodriguez, J.; Purnelle, M.; Episkopou, H.; Bertrand, L.; Francaux, M.; Deldicque, L.; Decottignies, A. Nuclear respiratory factor 1 and endurance exercise promote human telomere transcription. Sci. Adv. 2016, 2, e1600031. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.P.; Cifuentes-Rojas, C.; Kesner, B.; Aeby, E.; Lee, H.G.; Wei, C.; Oh, H.J.; Boukhali, M.; Haas, W.; Lee, J.T. TERRA RNA Antagonizes ATRX and Protects Telomeres. Cell 2017, 170, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Nergadze, S.G.; Farnung, B.O.; Wischnewski, H.; Khoriauli, L.; Vitelli, V.; Chawla, R.; Giulotto, E.; Azzalin, C.M. CpG-island promoters drive transcription of human telomeres. RNA 2009, 15, 2186–2194. [Google Scholar] [CrossRef] [PubMed]
- Riethman, H.; Ambrosini, A.; Paul, S. Human subtelomere structure and variation. Chromosome Res. 2005, 13, 505–515. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Almouzni, G. Assembly of telomeric chromatin to create ALTernative endings. Trends Cell Biol. 2014, 24, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Norseen, J.; Wiedmer, A.; Riethman, H.; Lieberman, P.M. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol. Cell 2009, 35, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Scheibe, M.; Arnoult, N.; Kappei, D.; Buchholz, F.; Decottignies, A.; Butter, F.; Mann, M. Quantitative interaction screen of telomeric repeat-containing RNA reveals novel TERRA regulators. Genome Res. 2013, 23, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Feuerhahn, S.; Lingner, J. TERRA-reinforced association of LSD1 with MRE11 promotes processing of uncapped telomeres. Cell Rep. 2014, 6, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Silanes, I.; Stagno d’Alcontres, M.; Blasco, M.A. TERRA transcripts are bound by a complex array of RNA-binding proteins. Nat. Commun. 2010, 1, 33. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Coen, D.M.; Knipe, D.M. Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. MBio 2013, 4, e00590–e00612. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, D.L.; Thompson, H.W.; Bloom, D.C. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J. Virol. 2009, 83, 8173–8181. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Ludlow, A.T.; Min, J.; Robin, J.D.; Stadler, G.; Mender, I.; Lai, T.P.; Zhang, N.; Wright, W.E.; Shay, J.W. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect-Over Long Distances (TPE-OLD): Implications for Aging and Cancer. PLoS Biol. 2016, 14, e2000016. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Wei, J.; Riethman, H.; Baur, J.A.; Voglauer, R.; Shay, J.W.; Wright, W.E. Telomere length regulates ISG15 expression in human cells. Aging 2009, 1, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Robin, J.D.; Ludlow, A.T.; Batten, K.; Magdinier, F.; Stadler, G.; Wagner, K.R.; Shay, J.W.; Wright, W.E. Telomere position effect: Regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 2014, 28, 2464–2476. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sandoval, A.; Gasser, S.M. On TADs and LADs: Spatial Control Over Gene Expression. Trends Genet. 2016, 32, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, Z.; Stong, N.; Plasschaert, R.; Moczan, A.; Chen, H.S.; Hu, S.; Wikramasinghe, P.; Davuluri, R.V.; Bartolomei, M.S.; et al. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J. 2012, 31, 4165–4178. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Keeping it quiet: Chromatin control of γ herpesvirus latency. Nat. Rev. Microbiol. 2013, 11, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Epigenetics and Genetics of Viral Latency. Cell Host Microbe 2016, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Zou, Y.; Shay, J.W.; Wright, W.E. Telomere position effect in human cells. Science 2001, 292, 2075–2077. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Weitzman, J.B. What’s the damage? The impact of pathogens on pathways that maintain host genome integrity. Cell Host Microbe 2014, 15, 283–294. [Google Scholar] [CrossRef] [PubMed]
- De Jong, R.N.; van der Vliet, P.C.; Brenkman, A.B. Adenovirus DNA replication: Protein priming, jumping back and the role of the DNA binding protein DBP. Curr. Top. Microbiol. Immunol. 2003, 272, 187–211. [Google Scholar] [PubMed]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, J.; Hammerschmidt, W. Structure and role of the terminal repeats of Epstein-Barr virus in processing and packaging of virion DNA. J. Virol. 1995, 69, 3147–3155. [Google Scholar] [PubMed]
- Kintner, C.R.; Sugden, B. The structure of the termini of the DNA of Epstein-Barr virus. Cell 1979, 17, 661–671. [Google Scholar] [CrossRef]
- Strang, B.L.; Stow, N.D. Circularization of the herpes simplex virus type 1 genome upon lytic infection. J. Virol. 2005, 79, 12487–12494. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.K.; Coen, D.M. Herpes simplex viruses: Mechanisms of DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a013011. [Google Scholar] [CrossRef] [PubMed]
- Shirata, N.; Kudoh, A.; Daikoku, T.; Tatsumi, Y.; Fujita, M.; Kiyono, T.; Sugaya, Y.; Isomura, H.; Ishizaki, K.; Tsurumi, T. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J. Biol. Chem. 2005, 280, 30336–30341. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.J.; Knipe, D.M. Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 2004, 78, 5856–5866. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, N.; Bai, P.; Buchek, G.; Korza, G.; Weller, S.K. Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J. Virol. 2010, 84, 12504–12514. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Carson, C.T.; Muotri, A.R.; Gage, F.H.; Weitzman, M.D. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 2005, 102, 5844–5849. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, G.S.; Durocher, D.; Weitzman, M.D. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Heming, J.D.; Huffman, J.B.; Jones, L.M.; Homa, F.L. Isolation and characterization of the herpes simplex virus 1 terminase complex. J. Virol. 2014, 88, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Marcomini, I.; Gasser, S.M. Nuclear organization in DNA end processing: Telomeres vs. double-strand breaks. DNA Repair (Amst.) 2015, 32, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Doksani, Y.; de Lange, T. The role of double-strand break repair pathways at functional and dysfunctional telomeres. Cold Spring Harb. Perspect. Biol. 2014, 6, a016576. [Google Scholar] [CrossRef] [PubMed]
- Darwish, A.S.; Grady, L.M.; Bai, P.; Weller, S.K. ICP8 Filament Formation Is Essential for Replication Compartment Formation during Herpes Simplex Virus Infection. J. Virol. 2015, 90, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Cardone, G.; Heymann, J.B.; Cheng, N.; Trus, B.L.; Steven, A.C. Procapsid assembly, maturation, nuclear exit: Dynamic steps in the production of infectious herpesvirions. Adv. Exp. Med. Biol. 2012, 726, 423–439. [Google Scholar] [PubMed]
- Scherer, M.; Stamminger, T. Emerging Role of PML Nuclear Bodies in Innate Immune Signaling. J. Virol. 2016, 90, 5850–5854. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. The use of fluorescence microscopy to study the association between herpesviruses and intrinsic resistance factors. Viruses 2011, 3, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Nagata, K.; Wodrich, H. The Role of Nuclear Antiviral Factors against Invading DNA Viruses: The Immediate Fate of Incoming Viral Genomes. Viruses 2016, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Murray, J. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 2005, 79, 5078–5089. [Google Scholar] [CrossRef] [PubMed]
- Cuchet-Lourenco, D.; Boutell, C.; Lukashchuk, V.; Grant, K.; Sykes, A.; Murray, J.; Orr, A.; Everett, R.D. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog. 2011, 7, e1002123. [Google Scholar] [CrossRef] [PubMed]
- Hannoun, Z.; Maarifi, G.; Chelbi-Alix, M.K. The implication of SUMO in intrinsic and innate immunity. Cytokine Growth Factor Rev. 2016, 29, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Cuchet-Lourenco, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011, 7, e1002245. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Nagata, K.; Wodrich, H. An Adenovirus DNA Replication Factor, but Not Incoming Genome Complexes, Targets PML Nuclear Bodies. J. Virol. 2015, 90, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Sindre, H.; Stamminger, T. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 2002, 76, 5769–5783. [Google Scholar] [CrossRef] [PubMed]
- Winkler, L.L.; Hwang, J.; Kalejta, R.F. Ubiquitin-independent proteasomal degradation of tumor suppressors by human cytomegalovirus pp71 requires the 19S regulatory particle. J. Virol. 2013, 87, 4665–4671. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.J.; Lieberman, P.M. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef] [PubMed]
- Grobelny, J.V.; Godwin, A.K.; Broccoli, D. ALT-associated PML bodies are present in viable cells and are enriched in cells in the G(2)/M phase of the cell cycle. J. Cell Sci. 2000, 113 Pt 24, 4577–4585. [Google Scholar] [PubMed]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar] [PubMed]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Xiang, T.; Pandita, T.K.; Gonzalez-Suarez, I.; Gonzalo, S.; Harris, C.C.; Yang, Q. Telomere recombination requires the MUS81 endonuclease. Nat. Cell Biol. 2009, 11, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Jiang, X.; Lee, W.H.; Chen, P.L. Assembly of functional ALT-associated promyelocytic leukemia bodies requires Nijmegen Breakage Syndrome 1. Cancer Res. 2003, 63, 2589–2595. [Google Scholar] [PubMed]
- Fan, Q.; Zhang, F.; Barrett, B.; Ren, K.; Andreassen, P.R. A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 2009, 37, 1740–1754. [Google Scholar] [CrossRef] [PubMed]
- Osterwald, S.; Deeg, K.I.; Chung, I.; Parisotto, D.; Worz, S.; Rohr, K.; Erfle, H.; Rippe, K. PML induces compaction, TRF2 depletion and DNA damage signaling at telomeres and promotes their alternative lengthening. J. Cell Sci. 2015, 128, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.; Leonhardt, H.; Rippe, K. De novo assembly of a PML nuclear subcompartment occurs through multiple pathways and induces telomere elongation. J. Cell Sci. 2011, 124 Pt 21, 3603–3618. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suva, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Kamranvar, S.A.; Chen, X.; Masucci, M.G. Telomere dysfunction and activation of alternative lengthening of telomeres in B-lymphocytes infected by Epstein-Barr virus. Oncogene 2013, 32, 5522–5530. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.K.; Sawitzke, J.A. Recombination promoted by DNA viruses: Phage lambda to herpes simplex virus. Annu. Rev. Microbiol. 2014, 68, 237–258. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, M.; Matocci, R.; Tasselli, L.; Cambiaghi, V.; Orleth, A.; Furia, L.; Marinelli, C.; Lombardi, S.; Sammarelli, G.; Aversa, F.; et al. PML is required for telomere stability in non-neoplastic human cells. Oncogene 2016, 35, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. IFI16 restricts HSV-1 replication by accumulating on the HSV-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 2014, 10, e1004503. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Diner, B.A.; Lum, K.K.; Cristea, I.M. The emerging role of nuclear viral DNA sensors. J. Biol. Chem. 2015, 290, 26412–26421. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Knipe, D.M. Cellular sensing of viral DNA and viral evasion mechanisms. Annu. Rev. Microbiol. 2014, 68, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Lengyel, P.; Liu, C.J. p204, a p200 family protein, as a multifunctional regulator of cell proliferation and differentiation. Cytokine Growth Factor Rev. 2008, 19, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Dutta, D.; Iqbal, J.; Pisano, G.; Gjyshi, O.; Ansari, M.A.; Kumar, B.; Chandran, B. Nuclear Innate Immune DNA Sensor IFI16 Is Degraded during Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV): Role of IFI16 in Maintenance of KSHV Latency. J. Virol. 2016, 90, 8822–8841. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Singh, V.V.; Dutta, S.; Veettil, M.V.; Dutta, D.; Chikoti, L.; Lu, J.; Everly, D.; Chandran, B. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J. Virol. 2013, 87, 8606–8623. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Kwon, K.M.; Kim, Y.E.; Kim, K.K.; Ahn, J.H. DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J. Virol. 2013, 87, 3076–3086. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef] [PubMed]
- Conrady, C.D.; Zheng, M.; Fitzgerald, K.A.; Liu, C.; Carr, D.J. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal. Immunol. 2012, 5, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Monack, D.M. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 2013, 13, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology 2015, 479–480, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Haronikova, L.; Coufal, J.; Kejnovska, I.; Jagelska, E.B.; Fojta, M.; Dvorakova, P.; Muller, P.; Vojtesek, B.; Brazda, V. IFI16 Preferentially Binds to DNA with Quadruplex Structure and Enhances DNA Quadruplex Formation. PLoS ONE 2016, 11, e0157156. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Kim, E.T.; Vladimirova, O.; Dheekollu, J.; Wang, Z.; Newhart, A.; Liu, D.; Myers, J.L.; Hensley, S.E.; Moffat, J.; et al. HSV-1 remodels host telomeres to facilitate viral replication. Cell Rep. 2014, 9, 2263–2278. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Roizman, B. The SP100 component of ND10 enhances accumulation of PML and suppresses replication and the assembly of HSV replication compartments. Proc. Natl. Acad. Sci. USA 2017, 114, E3823–E3829. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Mallon, S.; Roizman, B. PML plays both inimical and beneficial roles in HSV-1 replication. Proc. Natl. Acad. Sci. USA 2016, 113, E3022–E3028. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, P. The interaction between herpes simplex virus 1 genome and promyelocytic leukemia nuclear bodies (PML-NBs) as a hallmark of the entry in latency. Microb. Cell 2016, 3, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Catez, F.; Picard, C.; Held, K.; Gross, S.; Rousseau, A.; Theil, D.; Sawtell, N.; Labetoulle, M.; Lomonte, P. HSV-1 genome subnuclear positioning and associations with host-cell PML-NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog. 2012, 8, e1002852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caslini, C.; Connelly, J.A.; Serna, A.; Broccoli, D.; Hess, J.L. MLL associates with telomeres and regulates telomeric repeat-containing RNA transcription. Mol. Cell Biol. 2009, 29, 4519–4526. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Wang, X.; Campbell, M.R.; Song, L.; Safi, A.; Crawford, G.E.; Bell, D.A. Interactions of chromatin context, binding site sequence content, and sequence evolution in stress-induced p53 occupancy and transactivation. PLoS Genet. 2015, 11, e1004885. [Google Scholar] [CrossRef] [PubMed]
- Silva-Sousa, R.; Lopez-Panads, E.; Casacuberta, E. Drosophila telomeres: An example of co-evolution with transposable elements. Genome Dyn. 2012, 7, 46–67. [Google Scholar] [PubMed]
- Zhang, L.; Rong, Y.S. Retrotransposons at Drosophila telomeres: Host domestication of a selfish element for the maintenance of genome integrity. Biochim. Biophys. Acta 2012, 1819, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, L.; Lin, J.; Wang, A.; Wan, X.; Wu, Y.; Robson, S.C.; Sang, X.; Zhao, H. Distinct hepatitis B virus integration patterns in hepatocellular carcinoma and adjacent normal liver tissue. Int. J. Cancer 2017, 140, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Tutton, S.; Azzam, G.A.; Stong, N.; Vladimirova, O.; Wiedmer, A.; Monteith, J.A.; Beishline, K.; Wang, Z.; Deng, Z.; Riethman, H.; et al. Subtelomeric p53 binding prevents accumulation of DNA damage at human telomeres. EMBO J. 2016, 35, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lieberman, P.M. The crosstalk of telomere dysfunction and inflammation through cell-free TERRA containing exosomes. RNA Biol. 2016, 13, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Deng, Z.; Dahmane, N.; Tsai, K.; Wang, P.; Williams, D.R.; Kossenkov, A.V.; Showe, L.C.; Zhang, R.; Huang, Q.; et al. Telomeric repeat-containing RNA (TERRA) constitutes a nucleoprotein component of extracellular inflammatory exosomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6293–E6300. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [PubMed]
- Gutkin, A.; Uziel, O.; Beery, E.; Nordenberg, J.; Pinchasi, M.; Goldvaser, H.; Henick, S.; Goldberg, M.; Lahav, M. Tumor cells derived exosomes contain hTERT mRNA and transform nonmalignant fibroblasts into telomerase positive cells. Oncotarget 2016, 7, 59173–59188. [Google Scholar] [CrossRef] [PubMed]
- Al-Mayah, A.H.; Bright, S.J.; Bowler, D.A.; Slijepcevic, P.; Goodwin, E.; Kadhim, M.A. Exosome-Mediated Telomere Instability in Human Breast Epithelial Cancer Cells after X Irradiation. Radiat. Res. 2017, 187, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Baglio, S.R.; van Eijndhoven, M.A.; Koppers-Lalic, D.; Berenguer, J.; Lougheed, S.M.; Gibbs, S.; Leveille, N.; Rinkel, R.N.; Hopmans, E.S.; Swaminathan, S.; et al. Sensing of latent EBV infection through exosomal transfer of 5′pppRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E587–E596. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr virus encoded dUTPase containing exosomes modulate innate and adaptive immune responses in human dendritic cells and peripheral blood mononuclear cells. PLoS ONE 2013, 8, e69827. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Du, T.; Roizman, B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc. Natl. Acad. Sci. USA 2014, 111, E4991–E4996. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, S.; Kriss, M.; Golden-Mason, L.; Dobrinskikh, E.; Stone, A.E.; Soto-Gutierrez, A.; Mitchell, A.; Khetani, S.R.; Yamane, D.; Stoddard, M.; et al. Hepatitis C virus infection induces autocrine interferon signaling by human liver endothelial cells and release of exosomes, which inhibits viral replication. Gastroenterology 2015, 148, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Kadiu, I.; Narayanasamy, P.; Dash, P.K.; Zhang, W.; Gendelman, H.E. Biochemical and biologic characterization of exosomes and microvesicles as facilitators of HIV-1 infection in macrophages. J. Immunol. 2012, 189, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Jirintai, S.; Takahashi, M.; Kobayashi, T.; Nishizawa, T.; Kouki, T.; Yashiro, T.; Okamoto, H. Hepatitis E virus egress depends on the exosomal pathway, with secretory exosomes derived from multivesicular bodies. J. Gen. Virol. 2014, 95 Pt 10, 2166–2175. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, Y.; Huang, L. Exosomes from M1-Polarized Macrophages Potentiate the Cancer Vaccine by Creating a Pro-inflammatory Microenvironment in the Lymph Node. Mol. Ther. 2017, 25, 1665–1675. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Deng, Z.; Tutton, S.; Lieberman, P.M. The Telomeric Response to Viral Infection. Viruses 2017, 9, 218. https://doi.org/10.3390/v9080218
Wang Z, Deng Z, Tutton S, Lieberman PM. The Telomeric Response to Viral Infection. Viruses. 2017; 9(8):218. https://doi.org/10.3390/v9080218
Chicago/Turabian StyleWang, Zhuo, Zhong Deng, Steve Tutton, and Paul M. Lieberman. 2017. "The Telomeric Response to Viral Infection" Viruses 9, no. 8: 218. https://doi.org/10.3390/v9080218
APA StyleWang, Z., Deng, Z., Tutton, S., & Lieberman, P. M. (2017). The Telomeric Response to Viral Infection. Viruses, 9(8), 218. https://doi.org/10.3390/v9080218