
Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
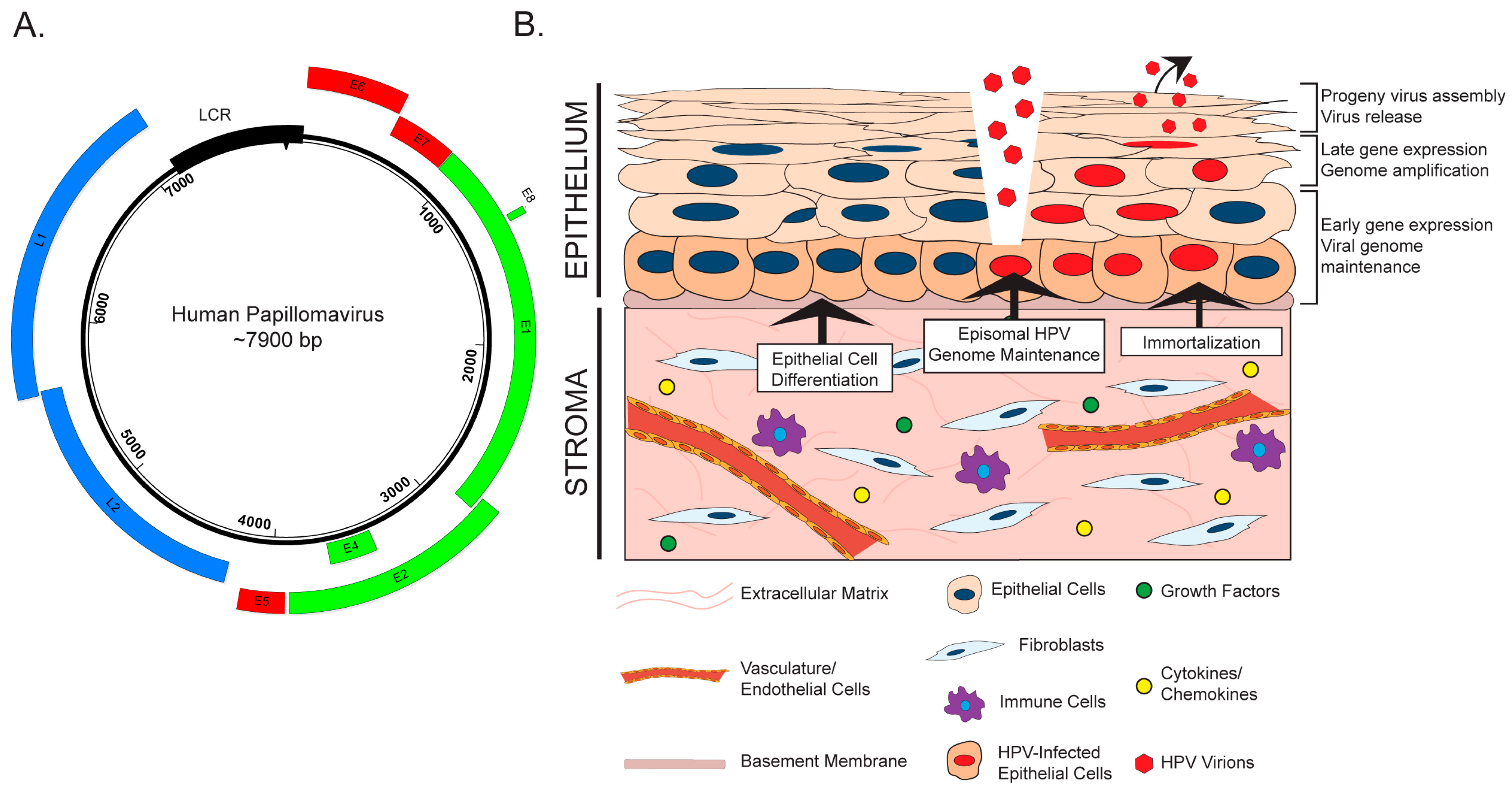
2. The HPV Life Cycle and the Stroma
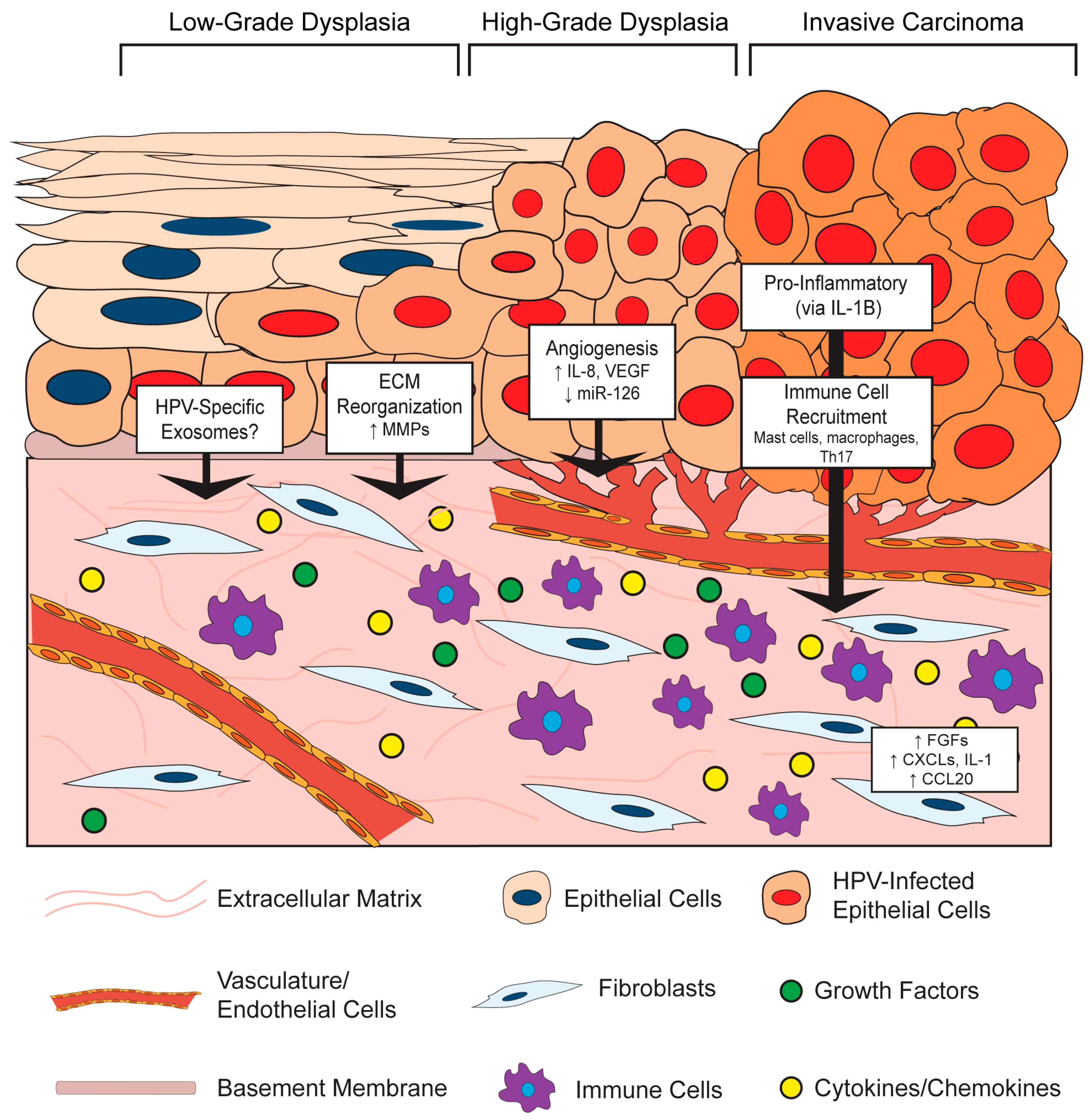
3. HPV and the Stroma: Bidirectional Paracrine Effects
3.1. Effects of the HPV-Positive Epithelium on the Stromal Microenvironment
3.1.1. HPV Effects on Stromal Architecture
3.1.2. HPV Effects on Angiogenesis in the Stroma
3.1.3. HPV Effects on Inflammation and Immune Cell Recruitment in the Stroma
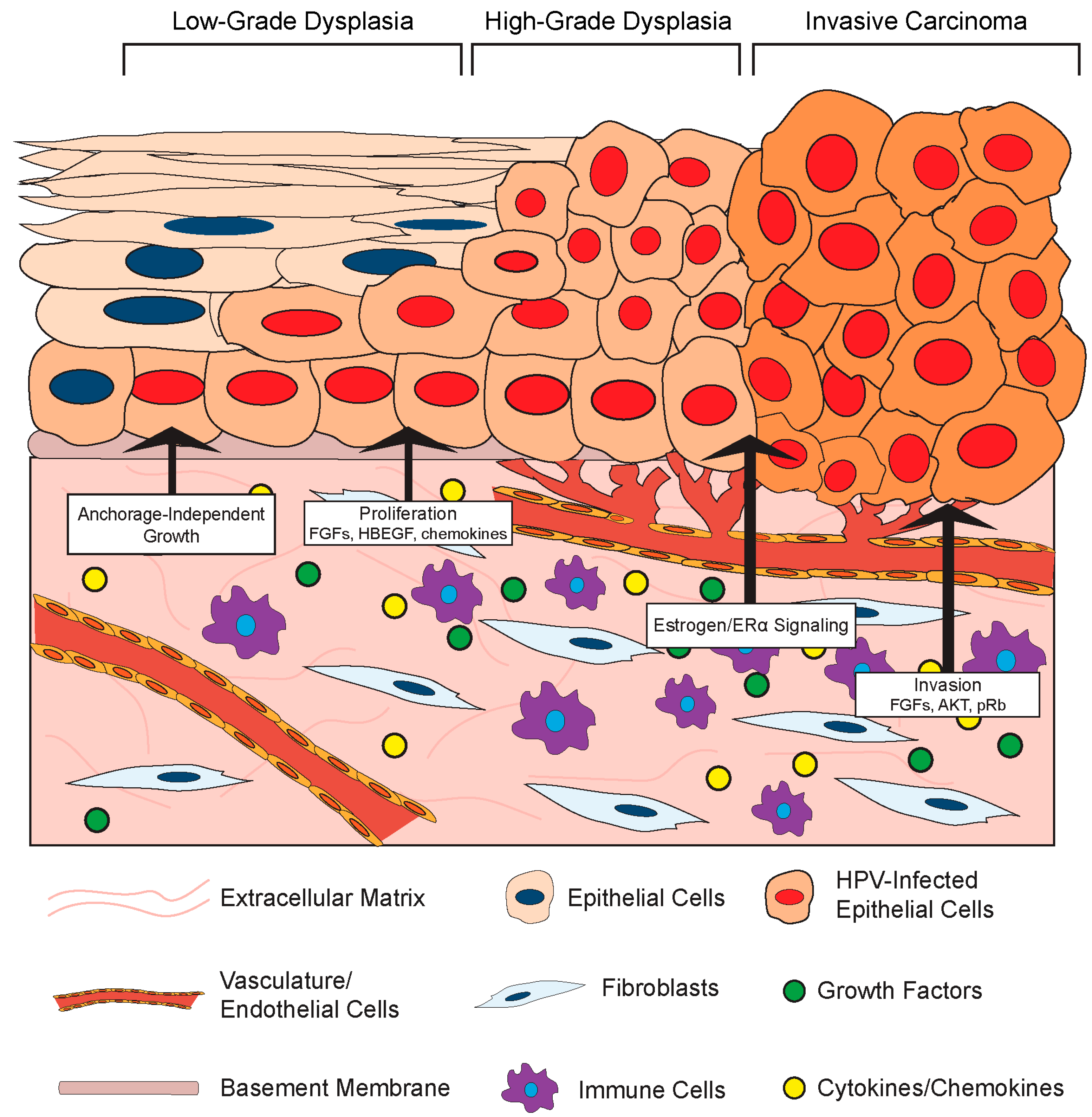
3.2. Effects of the Stromal Microenvironment on HPV-Positive Epithelia
3.2.1. Stromal Effects on Growth and Differentiation of HPV-Positive Epithelia
3.2.2. Stromal Effects on Disease in HPV-Associated Cancer Models
4. Potential Mechanisms of Bidirectional Crosstalk
5. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Mirvish, E.D.; Shuda, M. Strategies for human tumor virus discoveries: From microscopic observation to digital transcriptome subtraction. Front. Microbiol. 2016, 7, 676. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The papillomavirus episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, D499–D506. [Google Scholar] [CrossRef] [PubMed]
- Satterwhite, C.L.; Torrone, E.; Meites, E.; Dunne, E.F.; Mahajan, R.; Ocfemia, M.C.; Su, J.; Xu, F.; Weinstock, H. Sexually transmitted infections among us women and men: Prevalence and incidence estimates, 2008. Sex. Transm. Dis. 2013, 40, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Baseman, J.G.; Koutsky, L.A. The epidemiology of human papillomavirus infections. J. Clin. Virol. 2005, 32 (Suppl. S1), S16–S24. [Google Scholar] [CrossRef] [PubMed]
- Cogliano, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; WHO International Agency for Research on Cancer. Carcinogenicity of human papillomaviruses. Lancet Oncol. 2005, 6, 204. [Google Scholar] [CrossRef]
- D’Souza, G.; Kreimer, A.R.; Viscidi, R.; Pawlita, M.; Fakhry, C.; Koch, W.M.; Westra, W.H.; Gillison, M.L. Case-control study of human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007, 356, 1944–1956. [Google Scholar] [CrossRef] [PubMed]
- Hoots, B.E.; Palefsky, J.M.; Pimenta, J.M.; Smith, J.S. Human papillomavirus type distribution in anal cancer and anal intraepithelial lesions. Int. J. Cancer 2009, 124, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Bosch, F.X.; Castellsague, X.; Diaz, M.; de Sanjose, S.; Hammouda, D.; Shah, K.V.; Meijer, C.J. Against which human papillomavirus types shall we vaccinate and screen? The international perspective. Int. J. Cancer 2004, 111, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Frazer, I.H.; Leggatt, G.R.; Mattarollo, S.R. Prevention and treatment of papillomavirus-related cancers through immunization. Annu. Rev. Immunol. 2011, 29, 111–138. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Simard, E.P.; Dorell, C.; Noone, A.M.; Markowitz, L.E.; Kohler, B.; Eheman, C.; Saraiya, M.; Bandi, P.; Saslow, D.; et al. Annual report to the nation on the status of cancer, 1975–2009, featuring the burden and trends in human papillomavirus (HPV)-associated cancers and HPV vaccination coverage levels. J. Natl. Cancer Inst. 2013, 105, 175–201. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.C.; Schiffman, M.; Herrero, R.; Hildesheim, A.; Bratti, C.; Sherman, M.E.; Solomon, D.; Guillen, D.; Alfaro, M.; Morales, J.; et al. Longitudinal study of human papillomavirus persistence and cervical intraepithelial neoplasia grade 2/3: Critical role of duration of infection. J. Natl. Cancer Inst. 2010, 102, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Moscicki, A.B.; Schiffman, M.; Burchell, A.; Albero, G.; Giuliano, A.R.; Goodman, M.T.; Kjaer, S.K.; Palefsky, J. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine 2012, 30 (Suppl. S5), F24–F33. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Hernandez-Suarez, G.; Mendez, F.; Molano, M.; Posso, H.; Moreno, V.; Murillo, R.; Ronderos, M.; Meijer, C.; Munoz, A.; et al. Persistence of HPV infection and risk of high-grade cervical intraepithelial neoplasia in a cohort of colombian women. Br. J. Cancer 2009, 100, 1184–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffman, M.; Rodriguez, A.C.; Chen, Z.; Wacholder, S.; Herrero, R.; Hildesheim, A.; Desalle, R.; Befano, B.; Yu, K.; Safaeian, M.; et al. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 2010, 70, 3159–3169. [Google Scholar] [CrossRef] [PubMed]
- Luft, F.; Klaes, R.; Nees, M.; Durst, M.; Heilmann, V.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of integrated papillomavirus sequences by ligation-mediated PCR (DIPS-PCR) and molecular characterization in cervical cancer cells. Int. J. Cancer 2001, 92, 9–17. [Google Scholar] [CrossRef]
- Wentzensen, N.; Ridder, R.; Klaes, R.; Vinokurova, S.; Schaefer, U.; Doeberitz, M. Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene 2002, 21, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus genomes associate with BRD4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014, 10, e1004117. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Lambert, P.F. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: Implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Ziegert, C.; Wentzensen, N.; Vinokurova, S.; Kisseljov, F.; Einenkel, J.; Hoeckel, M.; von Knebel Doeberitz, M. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene 2003, 22, 3977–3984. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [PubMed]
- Munger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses causing cancer: Evasion from host-cell control in early events in carcinogenesis. J. Natl. Cancer Inst. 2000, 92, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Gius, D.; Funk, M.C.; Chuang, E.Y.; Feng, S.; Huettner, P.C.; Nguyen, L.; Bradbury, C.M.; Mishra, M.; Gao, S.; Buttin, B.M.; et al. Profiling microdissected epithelium and stroma to model genomic signatures for cervical carcinogenesis accommodating for covariates. Cancer Res. 2007, 67, 7113–7123. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M. Cross-roads in the classification of papillomaviruses. Virology 2013, 445, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.M.; Baker, C.C. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front. Biosci. 2006, 11, 2286–2302. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. S1), 2–23. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Lambert, P.F. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J. Virol. 2000, 74, 6622–6631. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Rheinwald, J.G.; Green, H. Serial cultivation of strains of human epidermal keratinocytes: The formation of keratinizing colonies from single cells. Cell 1975, 6, 331–343. [Google Scholar] [CrossRef]
- Llames, S.; Garcia-Perez, E.; Meana, A.; Larcher, F.; del Rio, M. Feeder layer cell actions and applications. Tissue Eng. Part B Rev. 2015, 21, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Dall, K.L.; Scarpini, C.G.; Roberts, I.; Winder, D.M.; Stanley, M.A.; Muralidhar, B.; Herdman, M.T.; Pett, M.R.; Coleman, N. Characterization of naturally occurring HPV16 integration sites isolated from cervical keratinocytes under noncompetitive conditions. Cancer Res. 2008, 68, 8249–8259. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lambert, P. Maintenance of HPV genomes in NOKs cells requires fibroblast feeder layer. McArdle Laboratory for Cancer Research, University of Wisconsin-Madison, Madison, WI, USA. Unpublished work. 2017. [Google Scholar]
- Werner, S.; Smola, H. Paracrine regulation of keratinocyte proliferation and differentiation. Trends Cell Biol. 2001, 11, 143–146. [Google Scholar] [CrossRef]
- Maas-Szabowski, N.; Shimotoyodome, A.; Fusenig, N.E. Keratinocyte growth regulation in fibroblast cocultures via a double paracrine mechanism. J. Cell Sci. 1999, 112 Pt 12, 1843–1853. [Google Scholar] [PubMed]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Laimins, L.A. In vitro systems for the study and propagation of human papillomaviruses. Curr. Top. Microbiol. Immunol. 1994, 186, 199–215. [Google Scholar] [PubMed]
- Lambert, P.F.; Ozbun, M.A.; Collins, A.; Holmgren, S.; Lee, D.; Nakahara, T. Using an immortalized cell line to study the HPV life cycle in organotypic “raft” cultures. Methods Mol. Med. 2005, 119, 141–155. [Google Scholar] [PubMed]
- Lee, D.; Norby, K.; Hayes, M.; Chiu, Y.F.; Sugden, B.; Lambert, P.F. Using organotypic epithelial tissue culture to study the human papillomavirus life cycle. Curr. Protoc. Microbiol. 2016, 41, 14B.8.1–14B.8.19. [Google Scholar] [PubMed]
- El Ghalbzouri, A.; Ponec, M. Diffusible factors released by fibroblasts support epidermal morphogenesis and deposition of basement membrane components. Wound Repair Regen. 2004, 12, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Maas-Szabowski, N.; Stark, H.J.; Fusenig, N.E. Keratinocyte growth regulation in defined organotypic cultures through IL-1-induced keratinocyte growth factor expression in resting fibroblasts. J. Investig. Dermatol. 2000, 114, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Smola, H.; Stark, H.J.; Thiekotter, G.; Mirancea, N.; Krieg, T.; Fusenig, N.E. Dynamics of basement membrane formation by keratinocyte-fibroblast interactions in organotypic skin culture. Exp. Cell Res. 1998, 239, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Smola, H.; Thiekotter, G.; Fusenig, N.E. Mutual induction of growth factor gene expression by epidermal-dermal cell interaction. J. Cell Biol. 1993, 122, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Schuster, C.; Rogon, Z.M.; Bauer, T.; Caushaj, N.; Baars, S.; Szabowski, S.; Bauer, C.; Schorpp-Kistner, M.; Hess, J.; et al. Efficient keratinocyte differentiation strictly depends on JNK-induced soluble factors in fibroblasts. J. Investig. Dermatol. 2014, 134, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte-fibroblast interactions in wound healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Schorey, J.S.; Harding, C.V. Extracellular vesicles and infectious diseases: New complexity to an old story. J. Clin. Investig. 2016, 126, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Pickard, A.; McDade, S.S.; McFarland, M.; McCluggage, W.G.; Wheeler, C.M.; McCance, D.J. HPV16 down-regulates the insulin-like growth factor binding protein 2 to promote epithelial invasion in organotypic cultures. PLoS Pathog. 2015, 11, e1004988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullar, A.; Dudas, J.; Olah, L.; Hollosi, P.; Papp, Z.; Sobel, G.; Karaszi, K.; Paku, S.; Baghy, K.; Kovalszky, I. Remodeling of extracellular matrix by normal and tumor-associated fibroblasts promotes cervical cancer progression. BMC Cancer 2015, 15, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Sakai, T.; Noguchi, Y.; Takita, M.; Hirakawa, S.; Ito, A. Tumor-stromal cell contact promotes invasion of human uterine cervical carcinoma cells by augmenting the expression and activation of stromal matrix metalloproteinases. Gynecol. Oncol. 2004, 92, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Smola-Hess, S.; Pahne, J.; Mauch, C.; Zigrino, P.; Smola, H.; Pfister, H.J. Expression of membrane type 1 matrix metalloproteinase in papillomavirus-positive cells: Role of the human papillomavirus (HPV) 16 and HPV8 E7 gene products. J. Gen. Virol. 2005, 86, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Kaewprag, J.; Umnajvijit, W.; Ngamkham, J.; Ponglikitmongkol, M. HPV16 oncoproteins promote cervical cancer invasiveness by upregulating specific matrix metalloproteinases. PLoS ONE 2013, 8, e71611. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Ye, M.; Zhang, W. E6/E7 oncoproteins of high risk HPV-16 upregulate MT1-MMP, MMP-2 and MMP-9 and promote the migration of cervical cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 4981–4989. [Google Scholar] [PubMed]
- Arbeit, J.M.; Munger, K.; Howley, P.M.; Hanahan, D. Progressive squamous epithelial neoplasia in K14-human papillomavirus type 16 transgenic mice. J. Virol. 1994, 68, 4358–4368. [Google Scholar] [PubMed]
- Herber, R.; Liem, A.; Pitot, H.; Lambert, P.F. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J. Virol. 1996, 70, 1873–1881. [Google Scholar] [PubMed]
- Song, S.; Pitot, H.C.; Lambert, P.F. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J. Virol. 1999, 73, 5887–5893. [Google Scholar] [PubMed]
- Lambert, P.F.; Pan, H.; Pitot, H.C.; Liem, A.; Jackson, M.; Griep, A.E. Epidermal cancer associated with expression of human papillomavirus type 16 E6 and E7 oncogenes in the skin of transgenic mice. Proc. Natl. Acad. Sci. USA 1993, 90, 5583–5587. [Google Scholar] [CrossRef] [PubMed]
- Strati, K.; Pitot, H.C.; Lambert, P.F. Identification of biomarkers that distinguish human papillomavirus (HPV)-positive versus HPV-negative head and neck cancers in a mouse model. Proc. Natl. Acad. Sci. USA 2006, 103, 14152–14157. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, M.K.; Pitot, H.C.; Liem, A.; Schweizer, J.; Mahoney, C.; Lambert, P.F. A mouse model for human anal cancer. Cancer Prev. Res. 2010, 3, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Hanahan, D.; Arbeit, J.M. Genetic predisposition and parameters of malignant progression in K14-HPV16 transgenic mice. Am. J. Pathol. 1996, 149, 1899–1917. [Google Scholar] [PubMed]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef] [PubMed]
- Smith-McCune, K.; Zhu, Y.H.; Hanahan, D.; Arbeit, J. Cross-species comparison of angiogenesis during the premalignant stages of squamous carcinogenesis in the human cervix and K14-HPV16 transgenic mice. Cancer Res. 1997, 57, 1294–1300. [Google Scholar] [PubMed]
- Mazibrada, J.; Ritta, M.; Mondini, M.; De Andrea, M.; Azzimonti, B.; Borgogna, C.; Ciotti, M.; Orlando, A.; Surico, N.; Chiusa, L.; et al. Interaction between inflammation and angiogenesis during different stages of cervical carcinogenesis. Gynecol. Oncol. 2008, 108, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, F.; Mead, L.; White, H.; Walker, J.; Ingram, D.A.; Roman, A. Human papillomavirus causes an angiogenic switch in keratinocytes which is sufficient to alter endothelial cell behavior. Virology 2007, 367, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ocejo, O.; Viloria-Petit, A.; Bequet-Romero, M.; Mukhopadhyay, D.; Rak, J.; Kerbel, R.S. Oncogenes and tumor angiogenesis: The HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene 2000, 19, 4611–4620. [Google Scholar] [CrossRef] [PubMed]
- Toussaint-Smith, E.; Donner, D.B.; Roman, A. Expression of human papillomavirus type 16 E6 and E7 oncoproteins in primary foreskin keratinocytes is sufficient to alter the expression of angiogenic factors. Oncogene 2004, 23, 2988–2995. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.; Smiley, L.C.; Ingram, D.; Roman, A. Expression of human papillomavirus type 16 E7 is sufficient to significantly increase expression of angiogenic factors but is not sufficient to induce endothelial cell migration. Virology 2011, 410, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Pilch, H.; Schlenger, K.; Steiner, E.; Brockerhoff, P.; Knapstein, P.; Vaupel, P. Hypoxia-stimulated expression of angiogenic growth factors in cervical cancer cells and cervical cancer-derived fibroblasts. Int. J. Gynecol. Cancer 2001, 11, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Chu, T.Y. Repression of mir-126 and upregulation of adrenomedullin in the stromal endothelium by cancer-stromal cross talks confers angiogenesis of cervical cancer. Oncogene 2014, 33, 3636–3647. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Gardiner, A.S.; Board, K.F.; Monzon, F.A.; Edwards, R.P.; Khan, S.A. Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene 2008, 27, 2575–2582. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Xie, H.; Zhou, Z.; Chen, H.; Hu, T.; Bai, Y.; Shen, Y.; Yuan, W.; Jing, Q.; et al. Endothelial-specific intron-derived miR-126 is down-regulated in human breast cancer and targets both VEGFA and PIK3R2. Mol. Cell. Biochem. 2011, 351, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine pdgf signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Kanodia, S.; Fahey, L.M.; Kast, W.M. Mechanisms used by human papillomaviruses to escape the host immune response. Curr. Cancer Drug Targets 2007, 7, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Mangino, G.; Chiantore, M.V.; Iuliano, M.; Fiorucci, G.; Romeo, G. Inflammatory microenvironment and human papillomavirus-induced carcinogenesis. Cytokine Growth Factor Rev. 2016, 30, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Woodby, B.; Scott, M.; Bodily, J. The interaction between human papillomaviruses and the stromal microenvironment. Prog. Mol. Biol. Transl. Sci. 2016, 144, 169–238. [Google Scholar] [PubMed]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. CXC chemokines in cancer angiogenesis and metastases. Adv. Cancer Res. 2010, 106, 91–111. [Google Scholar] [PubMed]
- Kumar, M.M.; Davuluri, S.; Poojar, S.; Mukherjee, G.; Bajpai, A.K.; Bafna, U.D.; Devi, U.K.; Kallur, P.P.; Kshitish, A.K.; Jayshree, R.S. Role of estrogen receptor α in human cervical cancer-associated fibroblasts: A transcriptomic study. Tumour Biol. 2016, 37, 4409–4420. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, M.E.; Horswill, M.; den Boon, J.A.; Barthakur, S.; Forouzan, O.; Beebe, D.J.; Roopra, A.; Ahlquist, P.; Lambert, P. Human papillomavirus oncogenes reprogram the cervical cancer microenvironment independently of and synergistically with estrogen. 2017; submitted for publication. [Google Scholar]
- Pahne-Zeppenfeld, J.; Schroer, N.; Walch-Ruckheim, B.; Oldak, M.; Gorter, A.; Hegde, S.; Smola, S. Cervical cancer cell-derived interleukin-6 impairs CCR7-dependent migration of MMP-9-expressing dendritic cells. Int. J. Cancer 2014, 134, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Schroer, N.; Pahne, J.; Walch, B.; Wickenhauser, C.; Smola, S. Molecular pathobiology of human cervical high-grade lesions: Paracrine STAT3 activation in tumor-instructed myeloid cells drives local MMP-9 expression. Cancer Res. 2011, 71, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Walch-Ruckheim, B.; Mavrova, R.; Henning, M.; Vicinus, B.; Kim, Y.J.; Bohle, R.M.; Juhasz-Boss, I.; Solomayer, E.F.; Smola, S. Stromal fibroblasts induce CCL20 through IL6/C/EBPβ to support the recruitment of Th17 cells during cervical cancer progression. Cancer Res. 2015, 75, 5248–5259. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.C.; Rossetti, R.A.; Lima, A.M.; Lepique, A.P. HPV associated tumor cells control tumor microenvironment and leukocytosis in experimental models. Immun. Inflamm. Dis. 2014, 2, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Lepique, A.P.; Daghastanli, K.R.; Cuccovia, I.M.; Villa, L.L. HPV16 tumor associated macrophages suppress antitumor T cell responses. Clin. Cancer Res. 2009, 15, 4391–4400. [Google Scholar] [CrossRef] [PubMed]
- Bolpetti, A.; Silva, J.S.; Villa, L.L.; Lepique, A.P. Interleukin-10 production by tumor infiltrating macrophages plays a role in human papillomavirus 16 tumor growth. BMC Immunol. 2010, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Tinkle, C.L.; Hanahan, D.; Werb, Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 2000, 103, 481–490. [Google Scholar] [CrossRef]
- Bergot, A.S.; Ford, N.; Leggatt, G.R.; Wells, J.W.; Frazer, I.H.; Grimbaldeston, M.A. HPV16-E7 expression in squamous epithelium creates a local immune suppressive environment via CCL2- and CCL5-mediated recruitment of mast cells. PLoS Pathog. 2014, 10, e1004466. [Google Scholar] [CrossRef] [PubMed]
- Choyce, A.; Yong, M.; Narayan, S.; Mattarollo, S.R.; Liem, A.; Lambert, P.F.; Frazer, I.H.; Leggatt, G.R. Expression of a single, viral oncoprotein in skin epithelium is sufficient to recruit lymphocytes. PLoS ONE 2013, 8, e57798. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Howley, P.M. Human papillomavirus immortalization and transformation functions. Virus Res. 2002, 89, 213–228. [Google Scholar] [CrossRef]
- Chapman, S.; McDermott, D.H.; Shen, K.; Jang, M.K.; McBride, A.A. The effect of Rho kinase inhibition on long-term keratinocyte proliferation is rapid and conditional. Stem Cell Res. Ther. 2014, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012, 180, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Quintero, J.; Baker, C.C. Keratinocyte growth conditions modulate telomerase expression, senescence, and immortalization by human papillomavirus type 16 E6 and E7 oncogenes. Cancer Res. 2003, 63, 7815–7824. [Google Scholar] [PubMed]
- Zheng, J.; Vaheri, A. Human skin fibroblasts induce anchorage-independent growth of HPV-16-DNA-immortalized cervical epithelial cells. Int. J. Cancer 1995, 61, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Maas-Szabowski, N.; Szabowski, A.; Stark, H.J.; Andrecht, S.; Kolbus, A.; Schorpp-Kistner, M.; Angel, P.; Fusenig, N.E. Organotypic cocultures with genetically modified mouse fibroblasts as a tool to dissect molecular mechanisms regulating keratinocyte growth and differentiation. J. Investig. Dermatol. 2001, 116, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Woodworth, C.D.; McMullin, E.; Iglesias, M.; Plowman, G.D. Interleukin 1α and tumor necrosis factor α stimulate autocrine amphiregulin expression and proliferation of human papillomavirus-immortalized and carcinoma-derived cervical epithelial cells. Proc. Natl. Acad. Sci. USA 1995, 92, 2840–2844. [Google Scholar] [CrossRef] [PubMed]
- Pickard, A.; Cichon, A.C.; Barry, A.; Kieran, D.; Patel, D.; Hamilton, P.; Salto-Tellez, M.; James, J.; McCance, D.J. Inactivation of Rb in stromal fibroblasts promotes epithelial cell invasion. EMBO J. 2012, 31, 3092–3103. [Google Scholar] [CrossRef] [PubMed]
- Pickard, A.; Cichon, A.C.; Menges, C.; Patel, D.; McCance, D.J. Regulation of epithelial differentiation and proliferation by the stroma: A role for the retinoblastoma protein. J. Investig. Dermatol. 2012, 132, 2691–2699. [Google Scholar] [CrossRef] [PubMed]
- Cichon, A.C.; Pickard, A.; McDade, S.S.; Sharpe, D.J.; Moran, M.; James, J.A.; McCance, D.J. AKT in stromal fibroblasts controls invasion of epithelial cells. Oncotarget 2013, 4, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Arbeit, J.M.; Olson, D.C.; Hanahan, D. Upregulation of fibroblast growth factors and their receptors during multi-stage epidermal carcinogenesis in K14-HPV16 transgenic mice. Oncogene 1996, 13, 1847–1857. [Google Scholar] [PubMed]
- Turner, M.A.; Darragh, T.; Palefsky, J.M. Epithelial-stromal interactions modulating penetration of matrigel membranes by HPV 16-immortalized keratinocytes. J. Investig. Dermatol. 1997, 109, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Mizushima, H.; Chinen, I.; Moribe, H.; Yagi, S.; Hoffman, R.M.; Kimura, T.; Yoshino, K.; Ueda, Y.; Enomoto, T.; et al. HB-EGF and PDGF mediate reciprocal interactions of carcinoma cells with cancer-associated fibroblasts to support progression of uterine cervical cancers. Cancer Res. 2011, 71, 6633–6642. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.F. Transgenic mouse models of tumor virus action. Annu. Rev. Virol. 2016, 3, 473–489. [Google Scholar] [CrossRef] [PubMed]
- Arbeit, J.M.; Howley, P.M.; Hanahan, D. Chronic estrogen-induced cervical and vaginal squamous carcinogenesis in human papillomavirus type 16 transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 2930–2935. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.R.; Duensing, S.; Brake, T.; Munger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003, 63, 4862–4871. [Google Scholar] [PubMed]
- Brake, T.; Lambert, P.F. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 2490–2495. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Wiedmeyer, K.; Shai, A.; Korach, K.S.; Lambert, P.F. Requirement for estrogen receptor alpha in a mouse model for human papillomavirus-associated cervical cancer. Cancer Res. 2008, 68, 9928–9934. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Lambert, P.F. Prevention and treatment of cervical cancer in mice using estrogen receptor antagonists. Proc. Natl. Acad. Sci. USA 2009, 106, 19467–19472. [Google Scholar] [CrossRef] [PubMed]
- Mehta, F.F.; Baik, S.; Chung, S.H. Recurrence of cervical cancer and its resistance to progestin therapy in a mouse model. Oncotarget 2017, 8, 2372–2380. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, M.E.; Chung, S.H.; Lambert, P.F. Recurrence of cervical cancer in mice after selective estrogen receptor modulator therapy. Am. J. Pathol. 2014, 184, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.A.; Son, J.; Mehta, F.F.; DeMayo, F.J.; Lydon, J.P.; Chung, S.H. Progesterone signaling inhibits cervical carcinogenesis in mice. Am. J. Pathol. 2013, 183, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Bronowicka-Klys, D.E.; Lianeri, M.; Jagodzinski, P.P. The role and impact of estrogens and xenoestrogen on the development of cervical cancer. Biomed. Pharmacother. 2016, 84, 1945–1953. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Franceschi, S.; Lambert, P.F. Estrogen and ERα: Culprits in cervical cancer? Trends Endocrinol. Metab. 2010, 21, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Mekada, E.; Hoffman, R.M. Reconstitution of a metastatic-resistant tumor microenvironment with cancer-associated fibroblasts enables metastasis. Cell Cycle 2017, 16, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Cottone, L.; Junankar, S.; Johansson, M.; DeNardo, D.G.; Korets, L.; Reinheckel, T.; Sloane, B.F.; Bogyo, M.; et al. Cathepsin C is a tissue-specific regulator of squamous carcinogenesis. Genes Dev. 2013, 27, 2086–2098. [Google Scholar] [CrossRef] [PubMed]
- Cooke, P.S.; Buchanan, D.L.; Young, P.; Setiawan, T.; Brody, J.; Korach, K.S.; Taylor, J.; Lubahn, D.B.; Cunha, G.R. Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc. Natl. Acad. Sci. USA 1997, 94, 6535–6540. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R. Stromal induction and specification of morphogenesis and cytodifferentiation of the epithelia of the mullerian ducts and urogenital sinus during development of the uterus and vagina in mice. J. Exp. Zool. 1976, 196, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R.; Cooke, P.S.; Kurita, T. Role of stromal-epithelial interactions in hormonal responses. Arch. Histol. Cytol. 2004, 67, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Kurita, T.; Cooke, P.S.; Cunha, G.R. Epithelial-stromal tissue interaction in paramesonephric (Mullerian) epithelial differentiation. Dev. Biol. 2001, 240, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Shin, M.K.; Korach, K.S.; Lambert, P.F. Requirement for stromal estrogen receptor α in cervical neoplasia. Horm. Cancer 2013, 4, 50–59. [Google Scholar] [CrossRef] [PubMed]
- den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [PubMed]
- Honegger, A.; Leitz, J.; Bulkescher, J.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Silencing of human papillomavirus (HPV) E6/E7 oncogene expression affects both the contents and the amounts of extracellular microvesicles released from HPV-positive cancer cells. Int. J. Cancer 2013, 133, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Chiantore, M.V.; Mangino, G.; Iuliano, M.; Zangrillo, M.S.; De Lillis, I.; Vaccari, G.; Accardi, R.; Tommasino, M.; Columba Cabezas, S.; Federico, M.; et al. Human papillomavirus E6 and E7 oncoproteins affect the expression of cancer-related microRNAs: Additional evidence in HPV-induced tumorigenesis. J. Cancer Res. Clin. Oncol. 2016, 142, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Harden, M.E.; Munger, K. Human papillomavirus 16 E6 and E7 oncoprotein expression alters microRNA expression in extracellular vesicles. Virology 2017, 508, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Honegger, A.; Schilling, D.; Bastian, S.; Sponagel, J.; Kuryshev, V.; Sultmann, H.; Scheffner, M.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Dependence of intracellular and exosomal micrornas on viral E6/E7 oncogene expression in HPV-positive tumor cells. PLoS Pathog. 2015, 11, e1004712. [Google Scholar] [CrossRef] [PubMed]
- Le Buanec, H.; D’Anna, R.; Lachgar, A.; Zagury, J.F.; Bernard, J.; Ittele, D.; d’Alessio, P.; Hallez, S.; Giannouli, C.; Burny, A.; et al. HPV-16 E7 but not E6 oncogenic protein triggers both cellular immunosuppression and angiogenic processes. Biomed. Pharmacother. 1999, 53, 424–431. [Google Scholar] [CrossRef]
- Le Buanec, H.; Lachgar, A.; D’Anna, R.; Zagury, J.F.; Bizzini, B.; Bernard, J.; Ittele, D.; Hallez, S.; Giannouli, C.; Burny, A.; et al. Induction of cellular immunosuppression by the human papillomavirus type 16 E7 oncogenic protein. Biomed. Pharmacother. 1999, 53, 323–328. [Google Scholar] [CrossRef]
- D’Anna, R.; Le Buanec, H.; Alessandri, G.; Caruso, A.; Burny, A.; Gallo, R.; Zagury, J.F.; Zagury, D.; D’Alessio, P. Selective activation of cervical microvascular endothelial cells by human papillomavirus 16-E7 oncoprotein. J. Natl. Cancer Inst. 2001, 93, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Vinzon, S.E.; Braspenning-Wesch, I.; Muller, M.; Geissler, E.K.; Nindl, I.; Grone, H.J.; Schafer, K.; Rosl, F. Protective vaccination against papillomavirus-induced skin tumors under immunocompetent and immunosuppressive conditions: A preclinical study using a natural outbred animal model. PLoS Pathog. 2014, 10, e1003924. [Google Scholar] [CrossRef] [PubMed]
- Ingle, A.; Ghim, S.; Joh, J.; Chepkoech, I.; Bennett Jenson, A.; Sundberg, J.P. Novel laboratory mouse papillomavirus (MusPV) infection. Vet. Pathol. 2011, 48, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Uberoi, A.; Yoshida, S.; Frazer, I.H.; Pitot, H.C.; Lambert, P.F. Role of ultraviolet radiation in papillomavirus-induced disease. PLoS Pathog. 2016, 12, e1005664. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spurgeon, M.E.; Lambert, P.F. Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis. Viruses 2017, 9, 219. https://doi.org/10.3390/v9080219
Spurgeon ME, Lambert PF. Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis. Viruses. 2017; 9(8):219. https://doi.org/10.3390/v9080219
Chicago/Turabian StyleSpurgeon, Megan E., and Paul F. Lambert. 2017. "Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis" Viruses 9, no. 8: 219. https://doi.org/10.3390/v9080219
APA StyleSpurgeon, M. E., & Lambert, P. F. (2017). Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis. Viruses, 9(8), 219. https://doi.org/10.3390/v9080219