Pharmacokinetics, Tissue Distribution and Excretion of a Novel Diuretic (PU-48) in Rats

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animal Handling
2.3. LC-MS/MS Analysis and Method Validation
2.4. HPLC Analysis and Method Validation
2.5. Preparation of Plasma, Tissue, Urine, Feces and Bile Samples
2.6. Plasma Pharmacokinetics
2.7. Tissue Distribution Experiments
2.8. Excretion Experiments
2.9. Plasma Protein Binding Assay
2.10. Pharmacokinetic Parameters and Statistical Analysis
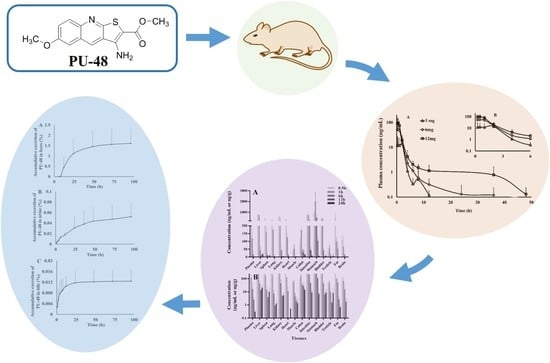
3. Results
3.1. Method Validation
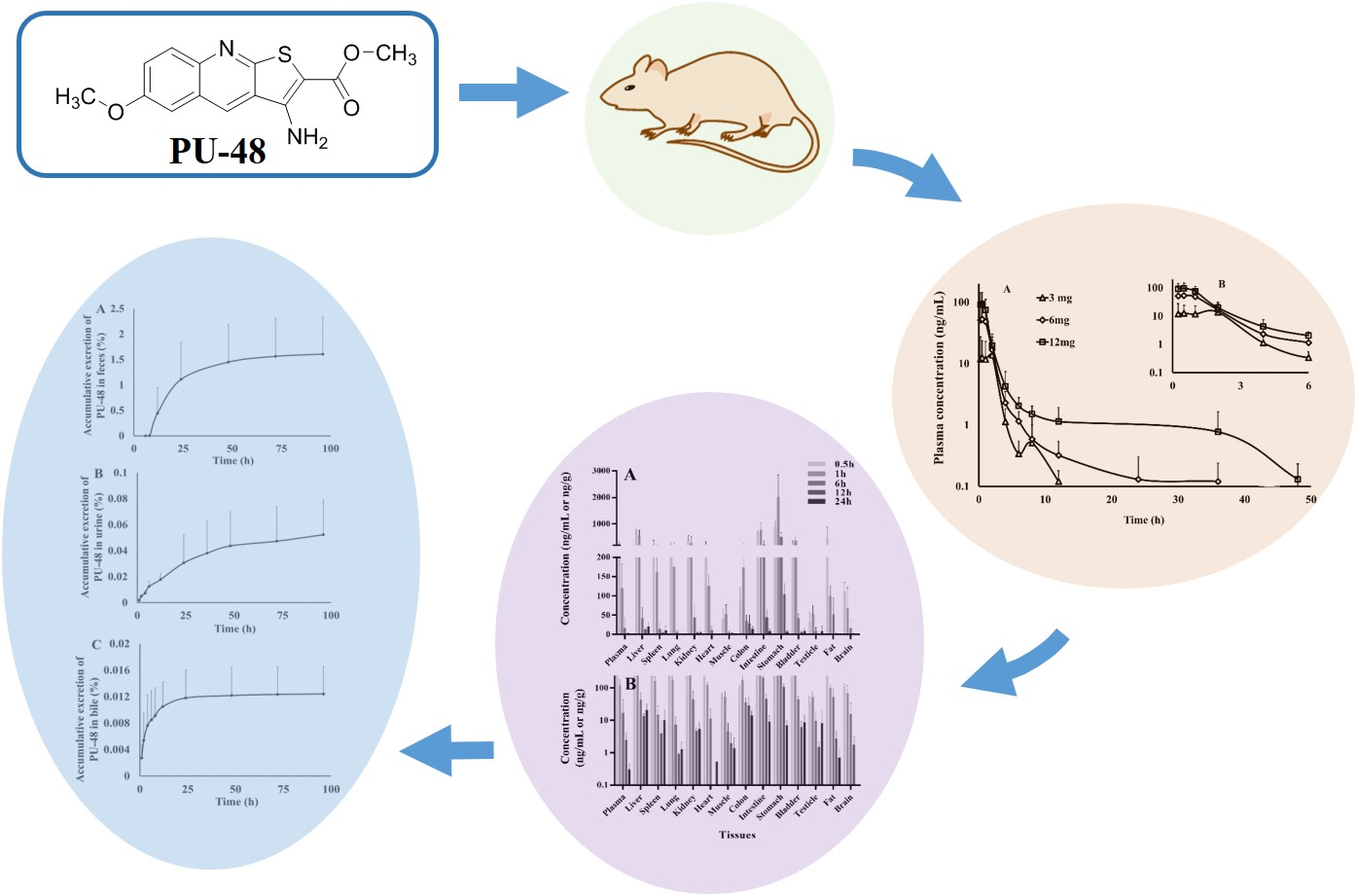
3.2. Pharmacokinetic Parameters
3.3. Tissue Distribution
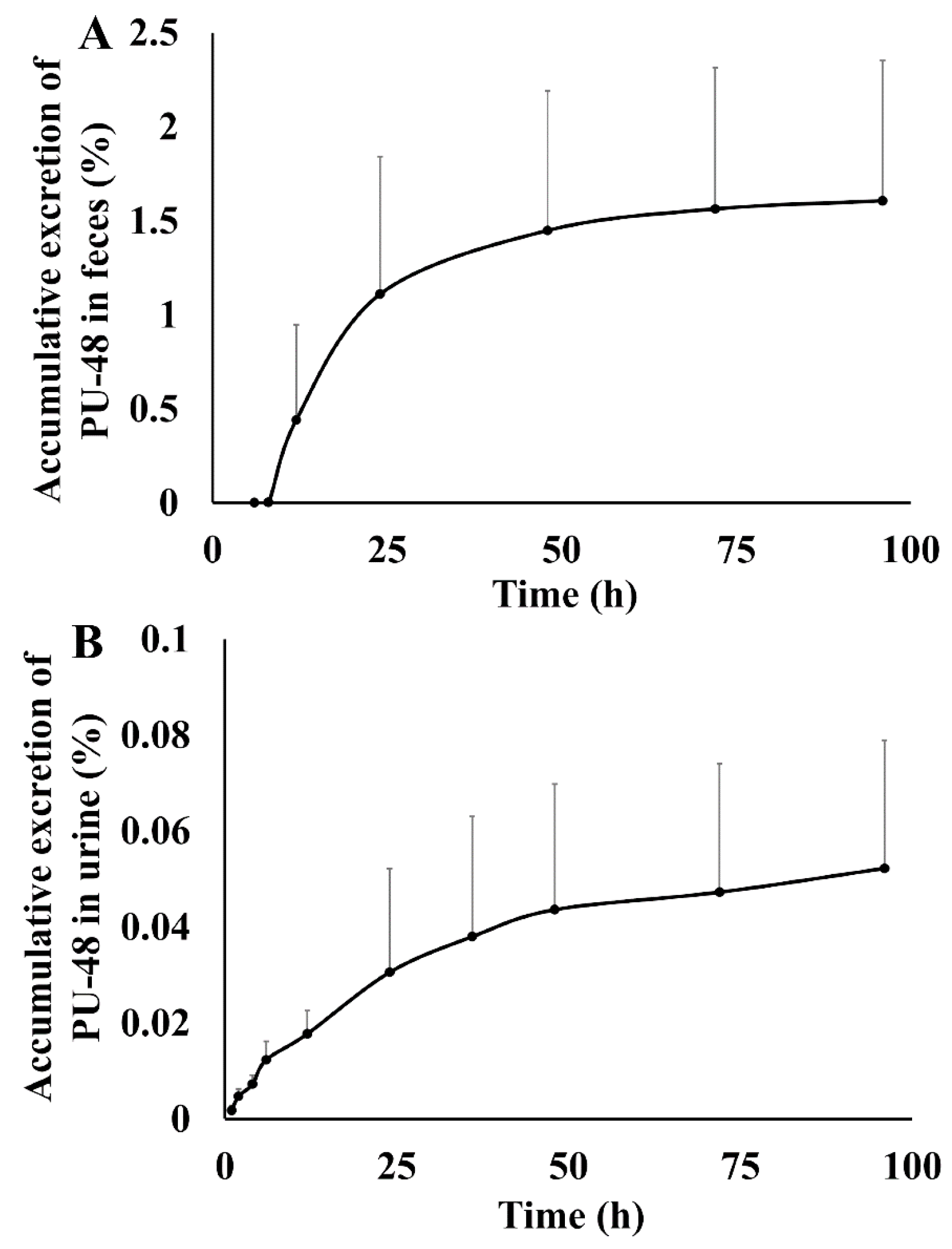
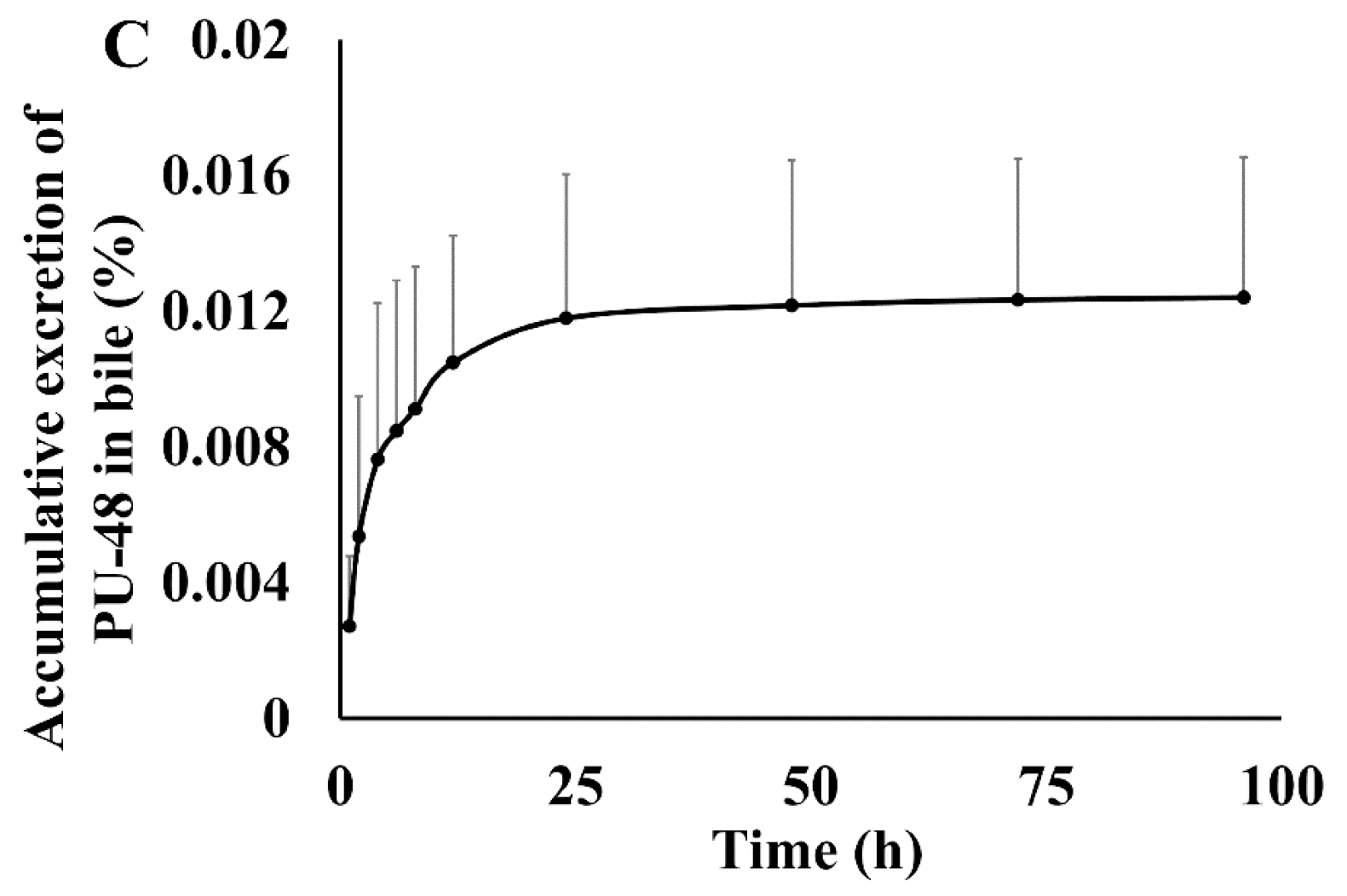
3.4. Excretion
3.5. Plasma Protein Binding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, F.; Lei, T.; Zhu, J.; Wang, W.; Sun, Y.; Chen, J.; Dong, Z.; Zhou, H.; Yang, B. A novel small-molecule thienoquinolin urea transporter inhibitor acts as a potential diuretic. Kidney Int. 2013, 83, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Wang, Y.; Xing, Y.; Ran, J.; Liu, M.; Lei, T.; Zhou, H.; Li, R.; Sands, J.M.; Yang, B. Thienoquinolins exert diuresis by strongly inhibiting UT-A urea transporters. Am. J. Physiol. Renal Physiol. 2014, 307, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Tou, W.I.; Zhou, H.; Li, F.; Ren, H.; Chen, C.Y.; Yang, B. Developing hypothetical inhibition mechanism of novel urea transporter B inhibitor. Sci. Rep. 2014, 4, 5775. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lau, C.W.; Jia, Y.; Li, Y.; Wang, W.; Ran, J.; Li, F.; Huang, Y.; Zhou, H.; Yang, B. Functional inhibition of urea transporter UT-B enhances endothelial-dependent vasodilatation and lowers blood pressure via l-arginine-endothelial nitric oxide synthase-nitric oxide pathway. Sci. Rep. 2016, 6, 18697. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.H.; de la Fuente, R.; Verkman, A.S. Urearetics: A small molecule screen yields nanomolar potency inhibitors of urea transporter UT-B. FASEB J. 2007, 21, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G. The emerging physiological roles of the SLC14A family of urea transporters. Br. J. Pharmacol. 2011, 164, 1780–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, J.D.; Sands, J.M. Urea transport and clinical potential of urearetics. Curr. Opin. Nephrol. Hypertens. 2016, 25, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Trinh-Trang-Tan, M.M.; Lasbennes, F.; Gane, P. UT-B1 proteins in rat: Tissue distribution and regulation by antidiuretic hormone in kidney. Am. J. Physiol. Renal Physiol. 2002, 283, F912–F922. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Zhou, L.; Layton, A.T.; Zhou, H.; Zhao, X.; Bankir, L.; Yang, B. Role of thin descending limb urea transport in renal urea handling and the urine concentrating mechanism. Am. J. Physiol. Renal Physiol. 2011, 301, F1251–F1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Bankir, L. Urea and urine concentrating ability: New insights from studies in mice. Am. J. Physiol. Renal Physiol. 2005, 288, F881–F896. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Bankir, L.; Gillespie, A.; Epstein, C.J.; Verkman, A.S. Urea-selective concentrating defect in transgenic mice lacking urea transporter UT-B. J. Biol. Chem. 2002, 277, 10633–10637. [Google Scholar] [CrossRef] [PubMed]
- Cafferkey, R.; Young, P.R.; McLaughlin, M.M.; Bergsma, D.J. Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol. Cell. Biol. 1993, 13, 6012–6023. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, G.; Yang, B. Urea transporter physiology studied in knockout mice. Front. Physiol. 2012, 3, 217. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Esteva-Font, C.; Cil, O.; Anderson, M.O.; Li, F.; Li, M.; Lei, T.; Ren, H.; Yang, B. Small-molecule inhibitors of urea transporters. Subcell. Biochem. 2014, 73, 165–177. [Google Scholar] [PubMed]
- Yang, B.; Verkman, A.S. Analysis of double knockout mice lacking aquaporin-1 and urea transporter UT-B. Evidence for UT-B-facilitated water transport in erythrocytes. J. Biol. Chem. 2002, 277, 36782–36786. [Google Scholar] [CrossRef] [PubMed]
- Bankir, L.; Chen, K.; Yang, B. Lack of UT-B in vasa recta and red blood cells prevents urea-induced improvement of urinary concentrating ability. Am. J. Physiol. Renal Physiol. 2004, 286, F144–F151. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Esteva-Font, C.; Yao, C.; Phuan, P.W.; Verkman, A.S.; Anderson, M.O. 1,1-Difluoroethyl-substituted triazolothienopyrimidines as inhibitors of a human urea transport protein (UT-B): New analogs and binding model. Bioorg. Med. Chem. Lett. 2013, 23, 3338–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sands, J.M. Urea transporter inhibitors: En route to new diuretics. Chem. Biol. 2013, 20, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Fenton, R.A.; Chou, C.L.; Stewart, G.S.; Smith, C.P.; Knepper, M.A. Urinary concentrating defect in mice with selective deletion of phloretin-sensitive urea transporters in the renal collecting duct. Proc. Natl. Acad. Sci. USA 2004, 101, 7469–7474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.; Anderson, M.O.; Zhang, J.; Yang, B.; Phuan, P.W.; Verkman, A.S. Triazolothienopyrimidine inhibitors of urea transporter UT-B reduce urine concentration. J. Am. Soc. Nephrol. 2012, 23, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Esteva-Font, C.; Phuan, P.W.; Anderson, M.O.; Verkman, A.S. A small molecule screen identifies selective inhibitors of urea transporter UT-A. Chem. Biol. 2013, 20, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Knepper, M.A.; Miranda, C.A. Urea channel inhibitors: A new functional class of aquaretics. Kidney Int. 2013, 83, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Kaji, D.M.; Diaz, J.; Parker, J.C. Urea inhibits Na-K-2Cl cotransport in medullary thick ascending limb cells. Am. J. Physiol. 1997, 272, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Pollare, T.; Lithell, H.; Berne, C. A comparison of the effects of hydrochlorothiazide and captopril on glucose and lipid metabolism in patients with hypertension. N. Engl. J. Med. 1989, 321, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Wang, X.; Liu, D.; Zhang, H.; Zhang, Q.; Lu, Y.Y.; Li, P.; Lou, Y.Q.; Yang, B.X.; Lu, C.; et al. Development and validation of an LC-MS/MS method for the determination of a novel thienoquinolin urea transporter inhibitor PU-48 in rat plasma and its application to a pharmacokinetic study. Biomed. Chromatogr. 2018, 32, e4157. [Google Scholar] [CrossRef] [PubMed]
- Wile, D. Diuretics: A review. Ann. Clin. Biochem. 2012, 49, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H.; Felker, G.M. Diuretic Treatment in Heart Failure. N. Engl. J. Med. 2017, 377, 1964–1975. [Google Scholar] [CrossRef] [PubMed]
- Kolber, M.R.; Garrison, S.; Turgeon, R.D. Electrolyte disturbance with diuretics and ACEIs. Can. Fam. Phys. 2016, 62, 569. [Google Scholar] [PubMed]
- Cheng, C.J.; Rodan, A.R.; Huang, C.L. Emerging Targets of Diuretic Therapy. Clin. Pharmacol. Ther. 2017, 102, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Esteva-Font, C.; Anderson, M.O.; Verkman, A.S. Urea transporter proteins as targets for small-molecule diuretics. Nat. Rev. Nephrol. 2015, 11, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, X.; Li, J.; Meng, Z.Y.; Li, S.C.; Li, Z.J.; Lu, Y.Y.; Ren, H.; Lou, Y.Q.; Lu, C.; et al. Quantitative and qualitative analysis of the novel antitumor 1,3,4-oxadiazole derivative (GLB) and its metabolites using HPLC-UV and UPLC-QTOF-MS. Sci. Rep. 2015, 5, 11906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, D.W.; Kim, Y.H.; Kim, H.H.; Ji, H.Y.; Yoo, S.D.; Choi, W.R.; Lee, S.M.; Han, C.K.; Lee, H.S. Dose-linear pharmacokinetics of oleanolic acid after intravenous and oral administration in rats. Biopharm. Drug Dispos. 2007, 28, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Wring, S.A.; Randolph, R.; Park, S.; Abruzzo, G.; Chen, Q.; Flattery, A.; Garrett, G.; Peel, M.; Outcalt, R.; Powell, K.; et al. Preclinical Pharmacokinetics and Pharmacodynamic Target of SCY-078, a First-in-Class Orally Active Antifungal Glucan Synthesis Inhibitor, in Murine Models of Disseminated Candidiasis. Antimicrob. Agents Chemother. 2017, 61, e02068-16. [Google Scholar] [CrossRef] [PubMed]
- Aylward, L.L.; Kirman, C.R.; Adgate, J.L.; McKenzie, L.M.; Hays, S.M. Interpreting variability in population biomonitoring data: Role of elimination kinetics. J. Expo. Sci. Environ. Epidemiol. 2012, 22, 398–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griesinger, G.; Dafopoulos, K.; Buendgen, N.; Cascorbi, I.; Georgoulias, P.; Zavos, A.; Messini, C.I.; Messinis, I.E. Elimination half-life of anti-Müllerian hormone. J. Clin. Endocrinol. Metab. 2012, 97, 2160–2163. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.Q.; Callegari, E.; Obach, R.S. The Use of In Vitro Data and Physiologically-Based Pharmacokinetic Modeling to Predict Drug Metabolite Exposure: Desipramine Exposure in Cytochrome P4502D6 Extensive and Poor Metabolizers Following Administration of Imipramine. Drug Metab. Dispos. 2016, 44, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Abel, S.; Russell, D.; Whitlock, L.A.; Ridgway, C.E.; Nedderman, A.N.; Walker, D.K. Assessment of the absorption, metabolism and absolute bioavailability of maraviroc in healthy male subjects. Br. J. Clin. Pharmacol. 2008, 65, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barve, A.; Chen, C.; Hebbar, V.; Desiderio, J.; Saw, C.L.; Kong, A.N. Metabolism, oral bioavailability and pharmacokinetics of chemopreventive kaempferol in rats. Biopharm. Drug Dispos. 2009, 30, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bu, F.; Li, L.; Jiao, Z.; Ma, G.; Cai, W.; Zhuang, X.; Lin, H.S.; Shin, J.G.; Xiang, X. Prediction of drug-drug interaction between Tacrolimus and principal ingredients of Wuzhi Capsule in Chinese healthy volunteers using physiologically-based pharmacokinetic modelling. Basic Clin. Pharmacol. Toxicol. 2018, 122, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Baptista, A.P.; Tamoutounour, S.; Zhuang, L.; Bouladoux, N.; Martins, A.J.; Huang, Y.; Gerner, M.Y.; Belkaid, Y.; Germain, R.N. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature 2018, 554, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Betz, A.L.; Keep, R.F.; Beer, M.E.; Ren, X.D. Blood-brain barrier permeability and brain concentration of sodium, potassium, and chloride during focal ischemia. J. Cereb. Blood Flow Metab. 1994, 14, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.K.; Nagashima, T.; Kondoh, T.; Tamaki, N. The effects of the Na(+)/Ca(++) exchange blocker on osmotic blood-brain barrier disruption. Brain Res. 2001, 900, 157–162. [Google Scholar] [CrossRef]
- Tournier, N.; Saba, W.; Cisternino, S.; Peyronneau, M.A.; Damont, A.; Goutal, S.; Dubois, A.; Dollé, F.; Scherrmann, J.M.; Valette, H.; et al. Effects of selected OATP and/or ABC transporter inhibitors on the brain and whole-body distribution of glyburide. AAPS J. 2013, 15, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Kort, A.; Durmus, S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Brain and Testis Accumulation of Regorafenib is Restricted by Breast Cancer Resistance Protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1). Pharm. Res. 2015, 32, 2205–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, T.; Haruta, T.; Shirasaka, Y.; Tamai, I. Organic cation transporter-mediated renal secretion of ipratropium and tiotropium in rats and humans. Drug Metab. Dispos. 2011, 39, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Nakajima, M.; Hatakeyama, M.; Fukami, T.; Yokoi, T. Preparation of a specific monoclonal antibody against human UDP-glucuronosyltransferase (UGT) 1A9 and evaluation of UGT1A9 protein levels in human tissues. Drug Metab. Dispos. 2012, 40, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Nordell, P.; Svanberg, P.; Bird, J.; Grime, K. Predicting metabolic clearance for drugs that are actively transported into hepatocytes: Incubational binding as a consequence of in vitro hepatocyte concentration is a key factor. Drug Metab. Dispos. 2013, 41, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Ma, S.; Yue, Q.; Kim-Kang, H.; Yi, Y.; Ly, J.; Boggs, J.W.; Fettes, A.; McClory, A.; Deng, Y.; et al. Absorption, metabolism and excretion of cobimetinib, an oral MEK inhibitor, in rats and dogs. Xenobiotica 2017, 47, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Soon, G.H.; Seng, K.Y.; Li, J.; Lee, E.; Yong, E.L.; Goh, B.C.; Flexner, C.; Lee, L. Pharmacokinetic modeling of plasma and intracellular concentrations of raltegravir in healthy volunteers. Antimicrob. Agents Chemother. 2011, 55, 4090–4095. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.H.; Lim, C.P.; Chen, Q.; Tang, C.C.; Kong, S.T.; Ho, P.C. Evaluating the relationship between Vancomycin trough concentration and 24-hour area under the concentration-time curve in neonates. Antimicrob. Agents Chemother. 2018, 62, pii:e01647-17. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.J.; Fan, J.; Chen, S.; Liu, L.; Sun, H.; Pang, K.S. Metabolite Kinetics: The Segregated Flow Model for Intestinal and Whole Body Physiologically Based Pharmacokinetic Modeling to Describe Intestinal and Hepatic Glucuronidation of Morphine in Rats In Vivo. Drug Metab. Dispos. 2016, 44, 1123–1138. [Google Scholar] [CrossRef] [PubMed]
- Samant, S.; Jiang, X.L.; Peletier, L.A.; Shuldiner, A.R.; Horenstein, R.B.; Lewis, J.P.; Lesko, L.J.; Schmidt, S. Identifying clinically relevant sources of variability: The clopidogrel challenge. Clin. Pharmacol. Ther. 2017, 101, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Chhonker, Y.S.; Chandasana, H.; Mukkavilli, R.; Prasad, Y.D.; Laxman, T.S.; Vangala, S.; Bhatta, R.S. Assessment of in vitro metabolic stability, plasma protein binding, and pharmacokinetics of E- and Z-guggulsterone in rat. Drug Test. Anal. 2016, 8, 966–975. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacokinetic Parameters | Unit | PU-48 | ||
|---|---|---|---|---|
| 3 mg/kg | 6 mg/kg | 12 mg/kg | ||
| Cmax | ng/mL | 12.6 ± 11.1 | 52.9 ± 46.8 | 94.3 ± 49.6 |
| Tmax | h | 1.19 ± 0.94 | 0.63 ± 0.31 | 0.50 ± 0.27 |
| AUC0–t | ng·h/mL | 60.0 ± 38.3 | 106.8 ± 53.0 | 178.9 ± 60.1 |
| AUC0–∞ | ng·h/mL | 60.5 ± 38.5 | 108.2 ± 52.5 | 180.7 ± 62.5 |
| t1/2 | h | 7.14 ± 2.93 | 7.00 ± 2.70 | 6.87 ± 3.49 |
| Tissues | Concentration of PU-48 (ng/mL or ng/g) | ||||
|---|---|---|---|---|---|
| 0.5 h | 1 h | 6 h | 12 h | 24 h | |
| Plasma | 276.7 ± 42.4 | 119.1 ± 63.5 | 16.8 ± 26.4 | 2.4 ± 1.7 | 0.3 ± 0.1 |
| Liver | 622.2 ± 182.8 | 534.3 ± 228.4 | 42.7 ± 27.3 | 13.4 ± 3.1 | 20.4 ± 12.2 |
| Spleen | 265.9 ± 120.1 | 161.2 ± 64.0 | 15.2 ± 12.9 | 3.8 ± 5.7 | 9.9 ± 12.0 |
| Lung | 231.0 ± 79.3 | 174.3 ± 91.4 | 7.2 ± 5.7 | 0.9 ± 0.1 | 1.3 ± 0.9 |
| Kidney | 415.8 ± 137.7 | 296.6 ± 241.4 | 44.5 ± 33.9 | 4.7 ± 1.1 | 5.3 ± 3.4 |
| Heart | 251.1 ± 78.4 | 124.6 ± 31.0 | 11.3 ± 11.2 | ND | 0.5 |
| Muscle | 39.8 ± 28.6 | 52.0 ± 25.5 | 4.5 ± 3.5 | 2.0 ± 1.9 | 1.4 ± 1.5 |
| Colon | 90.4 ± 30.0 | 172.5 ± 89.5 | 35.2 ± 14.7 | 28.6 ± 21.7 | 14.1 ± 5.6 |
| Intestine | 654.8 ± 89.3 | 777.4 ± 277.3 | 209.3 ± 166.5 | 45.4 ± 18.9 | 9.0 ± 4.7 |
| Stomach | 887.3 ± 234.9 | 2017.8 ± 821.5 | 508.8 ± 160.5 | 104.0 ± 28.4 | 7.0 ± 3.1 |
| Bladder | 258.2 ± 94.2 | 367.7 ± 123.8 | 43.4 ± 10.7 | 6.1 ± 3.2 | 8.6 ± 6.5 |
| Testicle | 34.2 ± 21.2 | 51.7 ± 24.4 | 9.5 ± 8.5 | 1.5 ± 0.7 | 1.4 ± 1.1 |
| Fat | 480.6 ± 398.1 | 99.3 ± 26.9 | 52.3 ± 43.2 | 2.7 ± 1.9 | 0.7 ± 0.6 |
| Brain | 111.1 ± 23.3 | 68.1 ± 52.7 | 15.7 ± 19.3 | 1.7 ± 1.5 | ND |
| Species | Added Concentration (µg/mL) | Concentration in Plasma (µg/mL) | Concentration in Buffer (µg/mL) | Protein-Binding Ratio (%) |
|---|---|---|---|---|
| Rat | 0.25 | 0.127 ± 0.007 | 0.012 ± 0.003 | 90.70 ± 2.18 |
| 1 | 0.524 ± 0.032 | 0.047 ± 0.002 | 91.06 ± 0.78 | |
| 4 | 2.128 ± 0.166 | 0.194 ± 0.019 | 90.83 ± 1.17 | |
| Human | 0.25 | 0.146 ± 0.008 | 0.012 ± 0.003 | 91.60 ± 1.57 |
| 1 | 0.611 ± 0.027 | 0.052 ± 0.003 | 91.48 ± 0.64 | |
| 4 | 2.290 ± 0.093 | 0.231 ± 0.029 | 89.90 ± 1.50 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-Y.; Zhang, H.; Liu, D.; Lu, Y.-Y.; Wang, X.; Li, P.; Lou, Y.-Q.; Yang, B.-X.; Lou, Y.-X.; Lu, C.; et al. Pharmacokinetics, Tissue Distribution and Excretion of a Novel Diuretic (PU-48) in Rats. Pharmaceutics 2018, 10, 124. https://doi.org/10.3390/pharmaceutics10030124
Zhang Z-Y, Zhang H, Liu D, Lu Y-Y, Wang X, Li P, Lou Y-Q, Yang B-X, Lou Y-X, Lu C, et al. Pharmacokinetics, Tissue Distribution and Excretion of a Novel Diuretic (PU-48) in Rats. Pharmaceutics. 2018; 10(3):124. https://doi.org/10.3390/pharmaceutics10030124
Chicago/Turabian StyleZhang, Zhi-Yuan, Hua Zhang, Dan Liu, Ying-Yuan Lu, Xin Wang, Pu Li, Ya-Qing Lou, Bao-Xue Yang, Ya-Xin Lou, Chuang Lu, and et al. 2018. "Pharmacokinetics, Tissue Distribution and Excretion of a Novel Diuretic (PU-48) in Rats" Pharmaceutics 10, no. 3: 124. https://doi.org/10.3390/pharmaceutics10030124
APA StyleZhang, Z. -Y., Zhang, H., Liu, D., Lu, Y. -Y., Wang, X., Li, P., Lou, Y. -Q., Yang, B. -X., Lou, Y. -X., Lu, C., Zhang, Q., & Zhang, G. -L. (2018). Pharmacokinetics, Tissue Distribution and Excretion of a Novel Diuretic (PU-48) in Rats. Pharmaceutics, 10(3), 124. https://doi.org/10.3390/pharmaceutics10030124