Metabolic Stability and Metabolite Characterization of Capilliposide B and Capilliposide C by LC–QTRAP–MS/MS


 and
and
Abstract
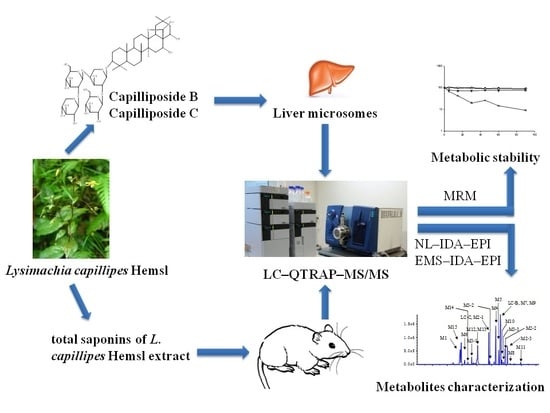
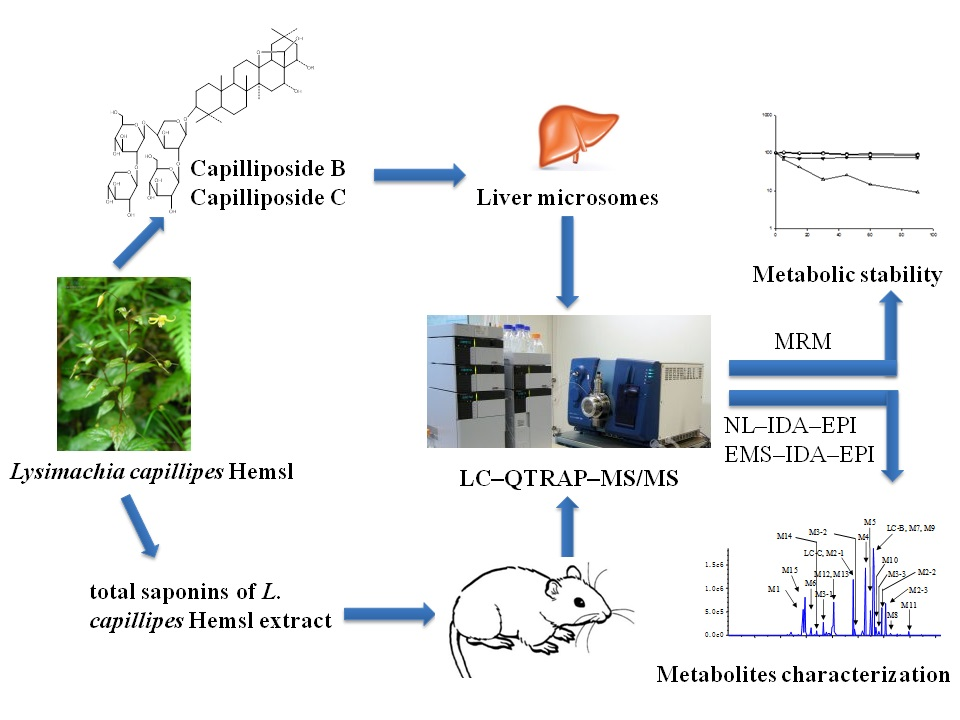
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
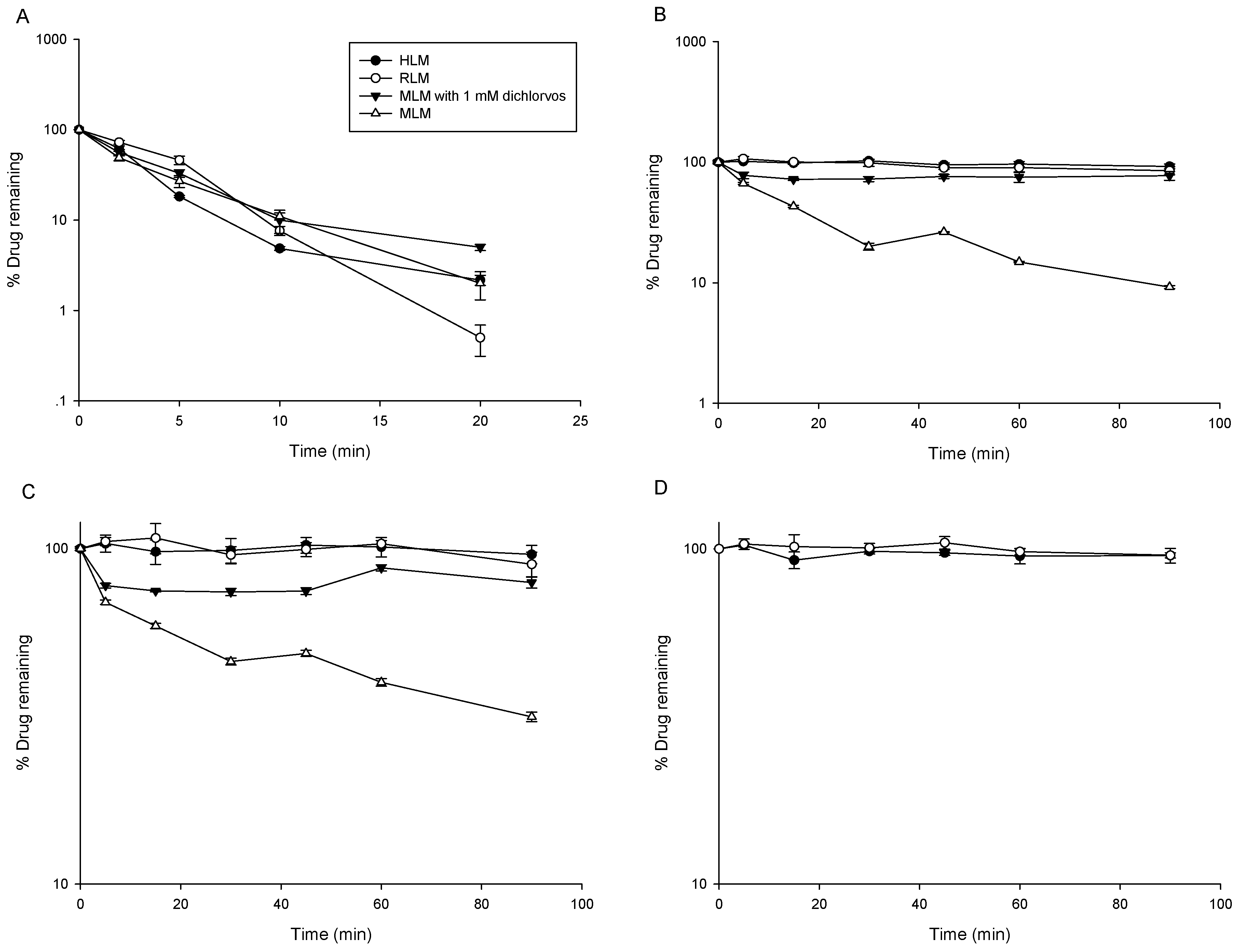
2.2. Metabolic Stability
2.3. Metabolism in Mice
2.3.1. Animal Experiments
2.3.2. Sample Preparation
2.3.3. LC-MS/MS Conditions
2.4. Data Analysis
3. Results and Discussion
3.1. Metabolic Stability
3.2. In Vivo Metabolism in Mice
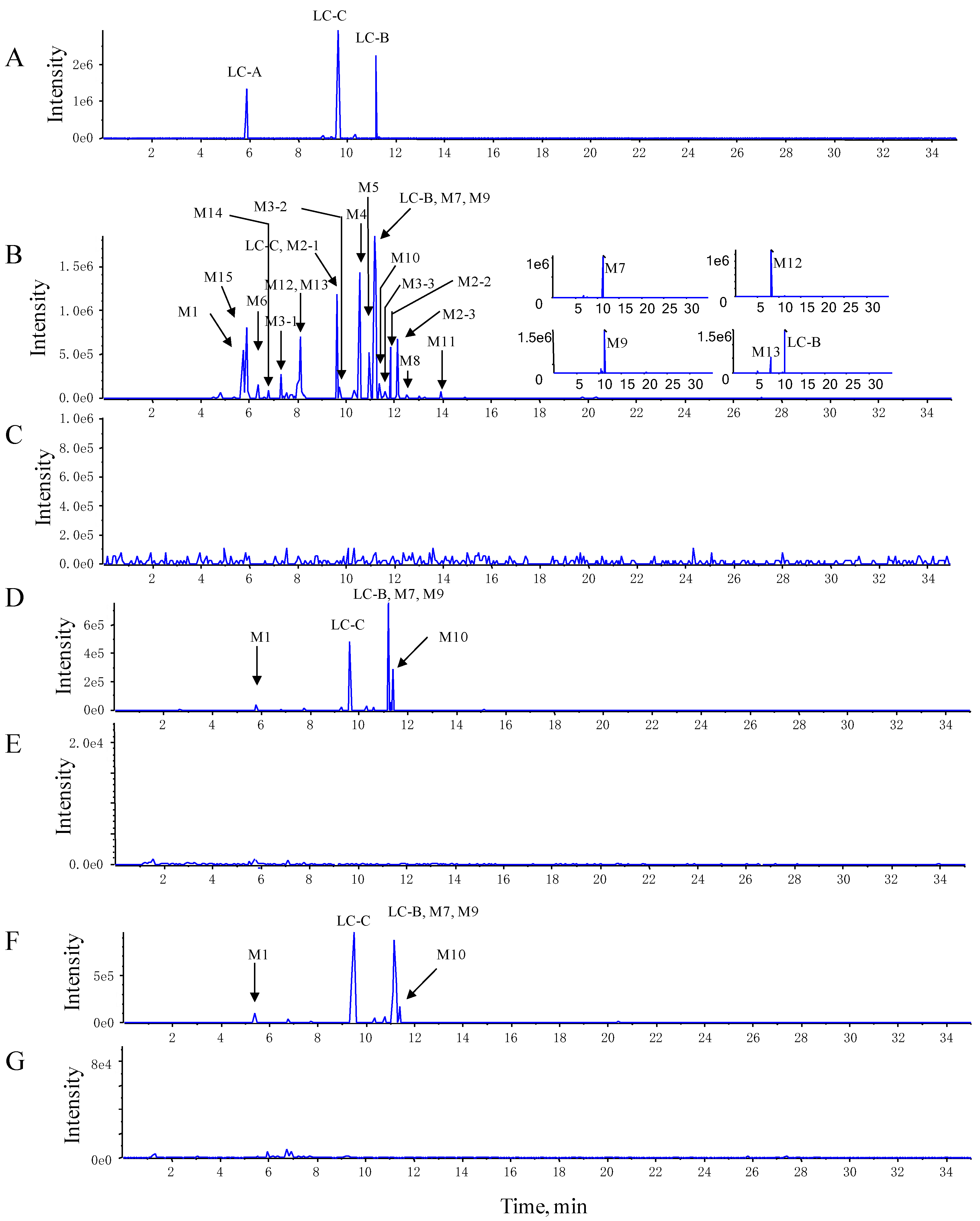
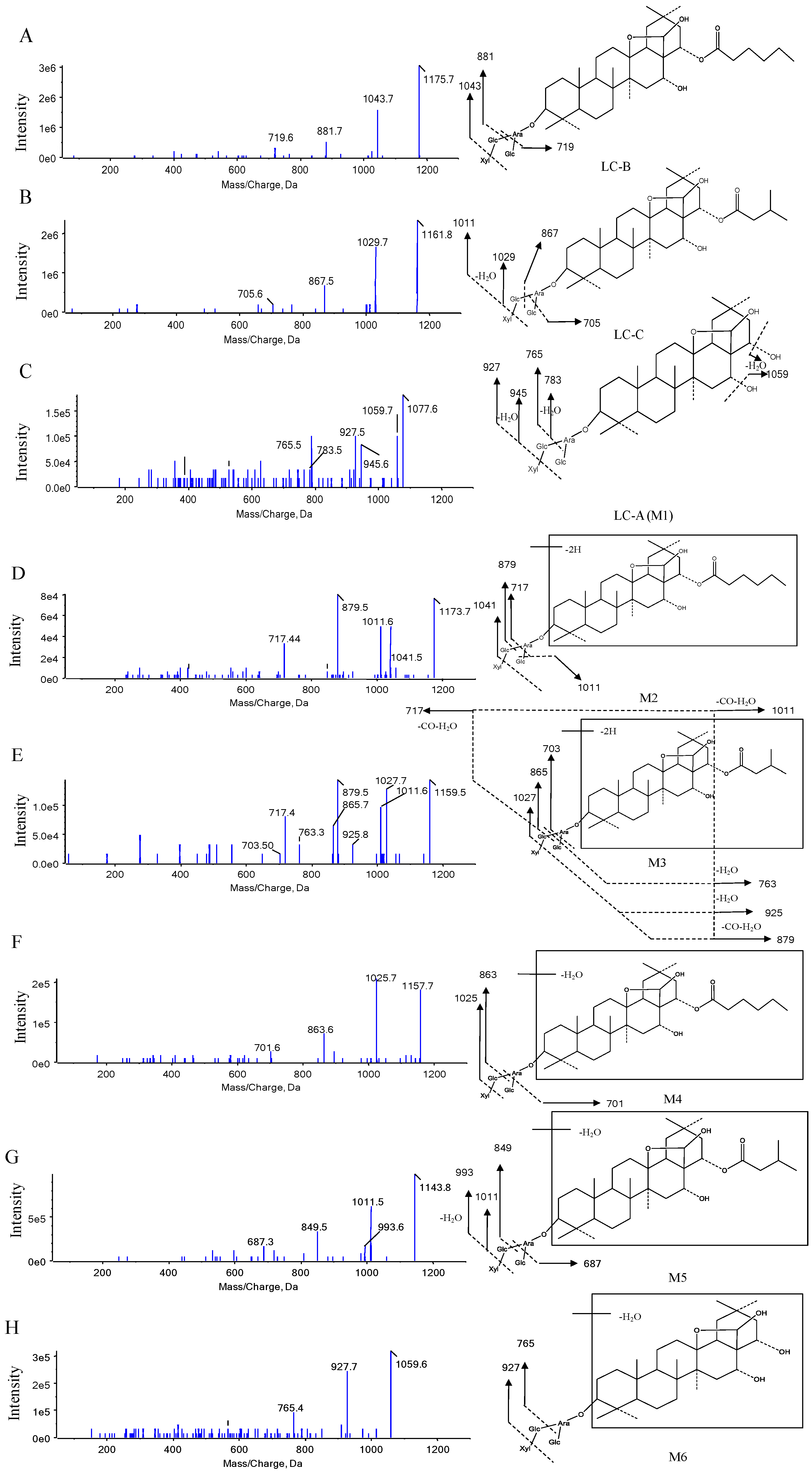
3.2.1. Metabolite Characterization by LC–QTRAP–MS/MS
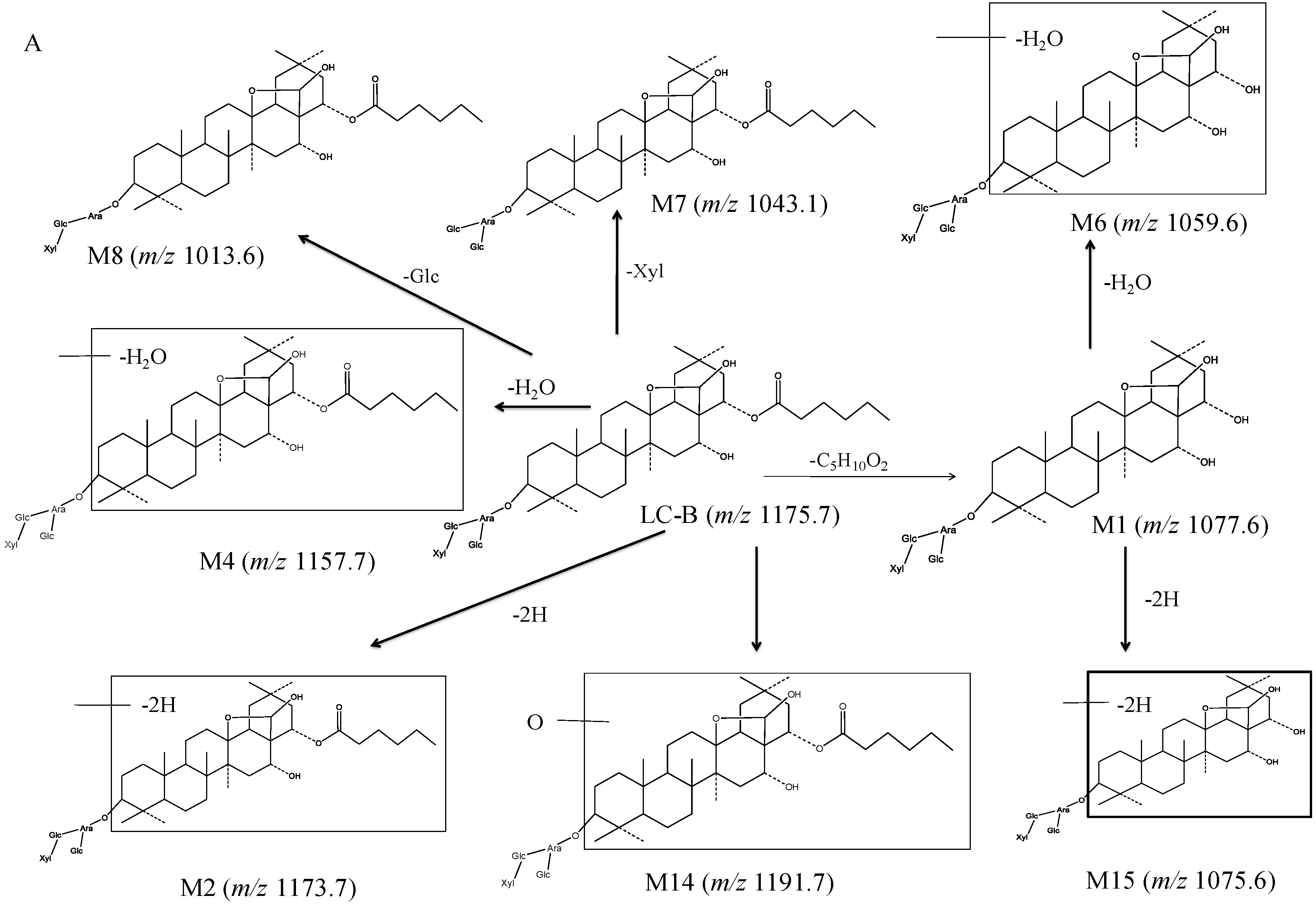
3.2.2. Proposed Metabolic Pathway of LC-B and LC-C in Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tian, J.K.; Xu, L.Z.; Zou, Z.M.; Yang, S.L. Two new triterpene saponins from Lysimachia capillipes. J. Asian Nat. Prod. Res. 2006, 8, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.K.; Xu, L.Z.; Zou, Z.M.; Yang, S.L. Three novel triterpenoid saponins from Lysimachia capillipes and their cytotoxic activities. Chem. Pharm. Bull. (Tokyo) 2006, 54, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Zhang, L.; Tian, J.; Xu, L.; Yang, S. Isolation and characterization of two new saponins from Lysimachia capillipes. Carbohydr. Res. 2006, 341, 2444–2448. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, L.; Zhang, L.; Chen, D.; Tian, J.; Cao, L.; Zhang, L. Capilliposide C derived from Lysimachia capillipes Hemsl inhibits growth of human prostate cancer PC3 cells by targeting caspase and MAPK pathways. Int. Urol. Nephrol. 2014, 46, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Xu, L.; Li, J.; Zhang, N. Capilliposide C Sensitizes Esophageal Squamous Carcinoma Cells to Oxaliplatin by Inducing Apoptosis Through the PI3K/Akt/mTOR Pathway. Med. Sci. Monit. 2017, 23, 2096–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, Z.H.; Wu, K.; Chen, Y.L.; Wang, B.; Zhang, S.R.; Ma, S.L. Capilliposide Isolated from Lysimachia capillipes Hemsl. Induces ROS Generation, Cell Cycle Arrest, and Apoptosis in Human Nonsmall Cell Lung Cancer Cell Lines. Evid. Based Complement. Altern. Med. 2014, 2014, 497456. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.U.; Rong, Y.M.; Ying, H.M.; Zou, L.B.; Tian, J.K. Experimental studies on anti-tumor effects of Capilliposide. Chin. Pharm. Bull. 2012, 28, 545–549. [Google Scholar]
- Yu, T.; Guo, D.; Wang, Y.; Zeng, J.; Chen, H.; Jiang, C.; Zheng, S. Study on GAP of Anti-cancer Drug Lysimachia capllipes Hemsl. Chin. Wild Plant. Resour. 2012, 3, 71–73. [Google Scholar]
- Cheng, Z.Z.; Zhang, L.; Zhang, Y.; Chen, G.Y.; Jiang, H.L. Simultaneous determination of capilliposide B and capilliposide C in rat plasma by LC-MS/MS and its application to a PK study. Bioanalysis. 2014, 6, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.Z.; Zhou, X.; Hu, B.Y.; Li, W.Y.; Chen, G.Y.; Zhang, Y.; Tian, J.K.; Zhang, L.; Li, M.; Jiang, H.L. Tissue distribution of capilliposide B, capilliposide C and their bioactive metabolite in mice using liquid-tandem mass spectrometry. Biomed. Chromatogr. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.Z.; Zhou, X.; Li, W.Y.; Hu, B.Y.; Zhang, Y.; Xu, Y.; Zhang, L.; Jiang, H.L. Optimization of solid-phase extraction and liquid chromatography-tandem mass spectrometry for simultaneous determination of capilliposide B and its active metabolite in rat urine and feces: Overcoming nonspecific binding. J. Pharm. Biomed. Anal. 2016, 131, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.Z.; Huang, M.L.; Chen, G.Y.; Yang, G.J.; Zhou, X.; Chen, C.; Zhang, Y.; Xu, Y.; Feng, Y.L.; Zhang, L.; et al. Cell-based assays in combination with ultra-high performance liquid chromatography-quadrupole time of flight tandem mass spectrometry for screening bioactive capilliposide C metabolites generated by rat intestinal microflora. J. Pharm. Biomed. Anal. 2016, 119, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.Z.; Ding, L.; Zhang, Y.; Chen, G.Y.; Chen, C.; Cheng, Y.Q.; Jiang, H.L. Determination of thailandepsin B in rat plasma with a LC-MS/MS method. Bioanalysis 2015, 7, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Berry, L.M.; Wollenberg, L.; Zhao, Z. Esterase activities in the blood, liver and intestine of several preclinical species and humans. Drug Metab. Lett. 2009, 3, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Zhu, M. Applications of mass spectrometry in drug metabolism: 50 years of progress. Drug Metab. Rev. 2015, 47, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Cheng, Z.Z.; Zhang, K.R.; Jiang, H.L.; Zhu, M.S. An integrated approach for profiling oxidative metabolites and glutathione adducts using liquid chromatography coupled with ultraviolet detection and triple quadrupole-linear ion trap mass spectrometry. J. Pharm. Biomed. Anal. 2016, 129, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Cheng, Z.Z.; Wang, L.; Feng, Y.L.; Huang, J.G.; Du, Z.F.; Jiang, H.L. A targeted strategy to identify untargeted metabolites from in vitro to in vivo: Rapid and sensitive metabolites profiling of licorice in rats using ultra-high performance liquid chromatography coupled with triple quadrupole-linear ion trap mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1092, 40–50. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Metabolic Description | Formula | Retention Time (min) | [M-H]− | Fragment Ions | Mouse | ||
|---|---|---|---|---|---|---|---|---|
| Plasma | Urine | Feces | ||||||
| LC-B | Parent | C58H96O24 | 11.20 | 1175.7 | 1043.7, 881.7, 719.6 | × | × | × |
| LC-C | Parent | C57H94O20 | 9.65 | 1161.8 | 1029.7, 1011.5, 867.5, 705.6 | × | × | × |
| M1 (LC-A) | Esterolysis | C52H86O23 | 5.88 | 1077.6 | 1059.7, 945.6, 927.5, 783.5, 765.5 | × | × | × |
| M2 | LC-B dehydrogenation | C58H94O24 | 9.73, 11.86, 12.13 | 1173.7 | 1041.5, 1011.6, 879.5, 717.4 | – | – | × |
| M3 | LC-C dehydrogenation | C57H92O20 | 7.68, 9.75, 11.41 | 1159.5 | 1027.6, 1011.7, 925.8, 879.5, 865.7, 763.3, 717.4, 703.5 | – | – | × |
| M4 | LC-B dehydration | C58H94O23 | 10.57 | 1157.7 | 1025.7, 863.6, 701.6 | – | − | × |
| M5 | LC-C dehydration | C57H92O23 | 10.98 | 1143.8 | 1011.5, 993.6, 849.5, 687.3 | – | – | × |
| M6 | LC-A dehydration | C52H84O22 | 6.39 | 1059.6 | 927.7, 765.4 | – | – | × |
| M7 | LC-B deglycosylation of xylose | C53H88O20 | 11.20 | 1043.1 | 881.3, 719.2 | × | × | × |
| M8 | LC-B deglycosylation of glucose | C52H86O19 | 12.21 | 1013.6 | 881.7, 719.4 | – | – | × |
| M9 | LC-C deglycosylation of xylose | C52H86O20 | 11.24 | 1029.5 | 867.5, 705.4 | × | × | × |
| M10 | LC-C deglycosylation of glucose | C51H84O19 | 11.39 | 999.6 | 897.6, 867.6, 765.4, 705.5, 659.5 | × | × | × |
| M11 | LC-C deglycosylation of glucose and xylose | C46H76O15 | 13.94 | 867.5 | 705.4 | – | – | × |
| M12 | LC-C hydroxylation | C58H98O24 | 8.18 | 1177.7 | 1045.7, 1027.8, 883.4 | – | – | × |
| M13 | LC-C methylation or hydroxylation with dehydrogenation | C58H96O24 | 8.13 | 1175.7 | 1043.7, 881.7, 719.6 | – | – | × |
| M14 | LC-B hydroxylation | C57H92O26 | 7.36 | 1191.7 | 1059.7, 897.7, 735.4 | – | – | × |
| M15 | LC-A dehydrogenation | C52H84O23 | 5.91 | 1075.6 | 943.6, 925.7, 879.7, 781.5, 763.4 | – | – | × |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Z.; Zhou, X.; Du, Z.; Li, W.; Hu, B.; Tian, J.; Zhang, L.; Huang, J.; Jiang, H. Metabolic Stability and Metabolite Characterization of Capilliposide B and Capilliposide C by LC–QTRAP–MS/MS. Pharmaceutics 2018, 10, 178. https://doi.org/10.3390/pharmaceutics10040178
Cheng Z, Zhou X, Du Z, Li W, Hu B, Tian J, Zhang L, Huang J, Jiang H. Metabolic Stability and Metabolite Characterization of Capilliposide B and Capilliposide C by LC–QTRAP–MS/MS. Pharmaceutics. 2018; 10(4):178. https://doi.org/10.3390/pharmaceutics10040178
Chicago/Turabian StyleCheng, Zhongzhe, Xing Zhou, Zhifeng Du, Wenyi Li, Bingying Hu, Jingkui Tian, Lin Zhang, Jiangeng Huang, and Hongliang Jiang. 2018. "Metabolic Stability and Metabolite Characterization of Capilliposide B and Capilliposide C by LC–QTRAP–MS/MS" Pharmaceutics 10, no. 4: 178. https://doi.org/10.3390/pharmaceutics10040178
APA StyleCheng, Z., Zhou, X., Du, Z., Li, W., Hu, B., Tian, J., Zhang, L., Huang, J., & Jiang, H. (2018). Metabolic Stability and Metabolite Characterization of Capilliposide B and Capilliposide C by LC–QTRAP–MS/MS. Pharmaceutics, 10(4), 178. https://doi.org/10.3390/pharmaceutics10040178