A Review on Liquid Chromatography-Tandem Mass Spectrometry Methods for Rapid Quantification of Oncology Drugs


 ,
, 

Abstract
:1. Introduction
2. Method of Literature Search
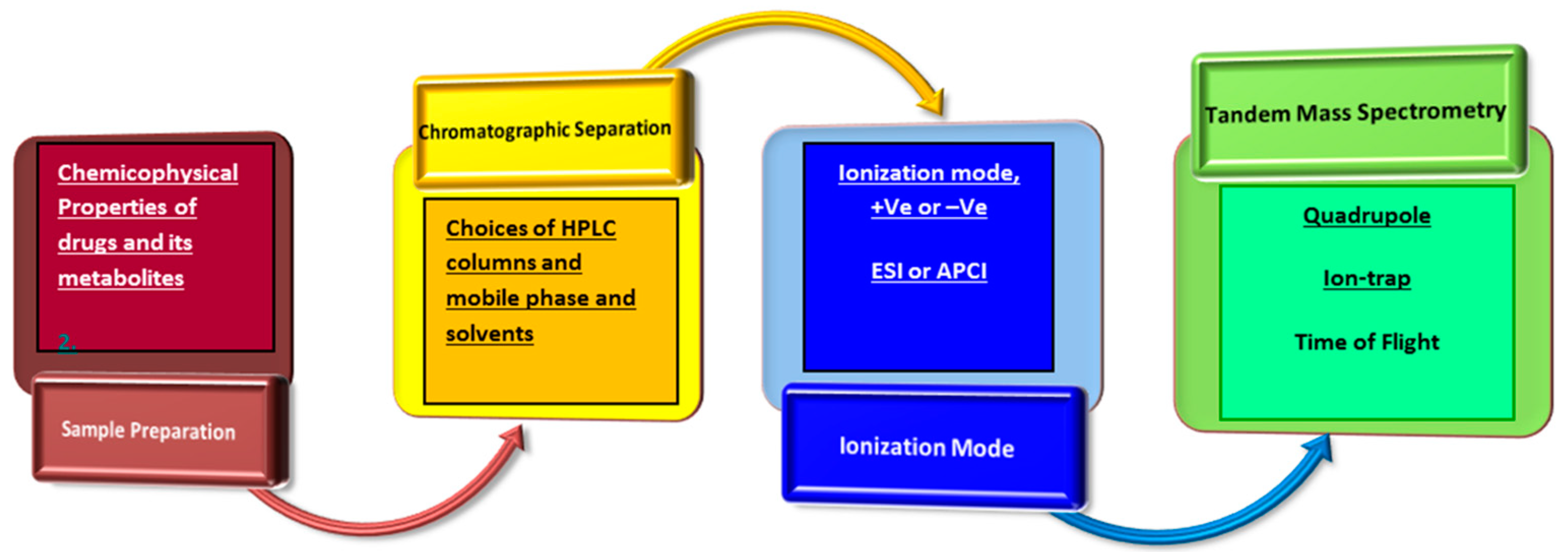
3. Results and Discussion
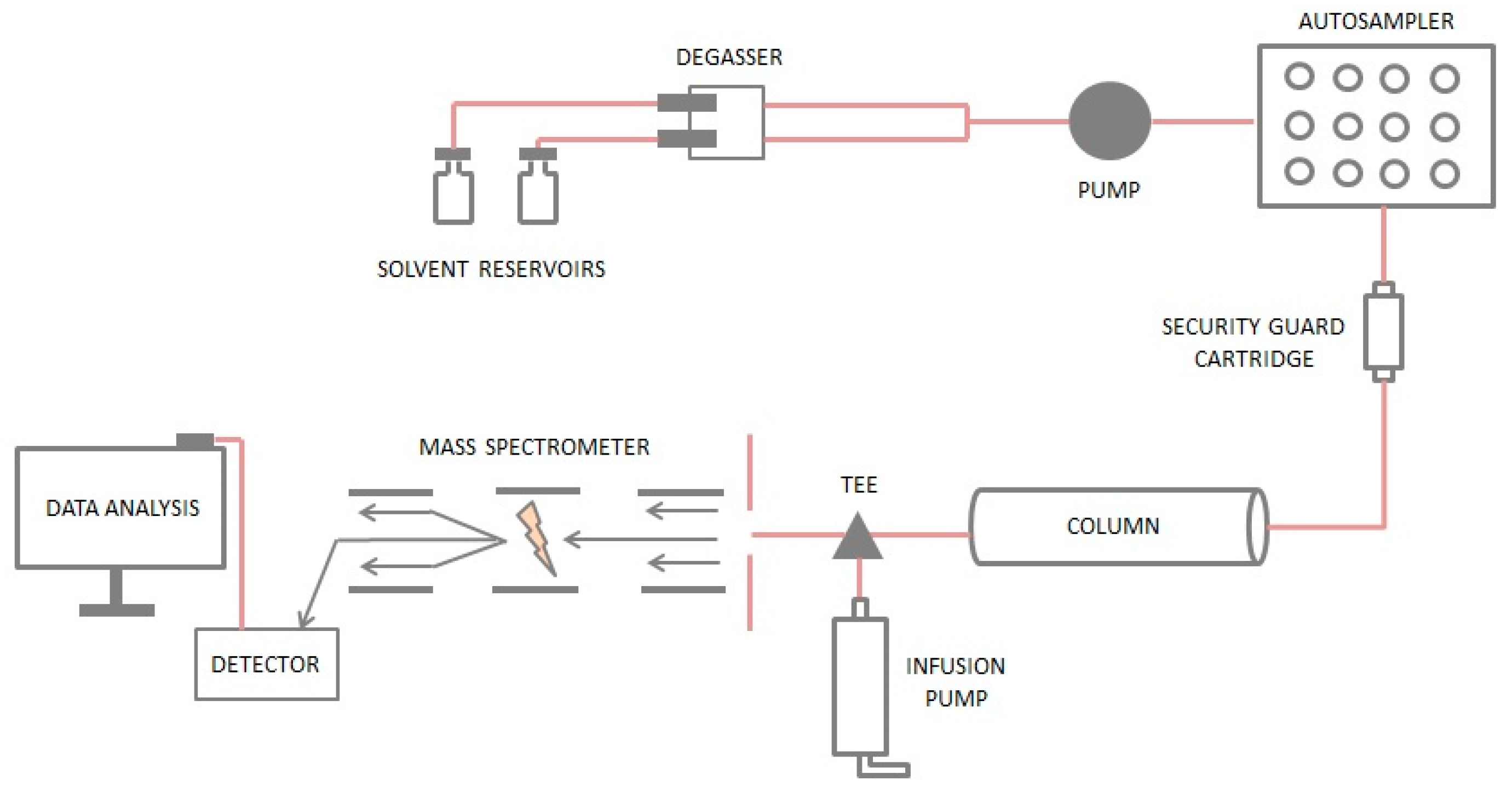
3.1. Sample Preparation
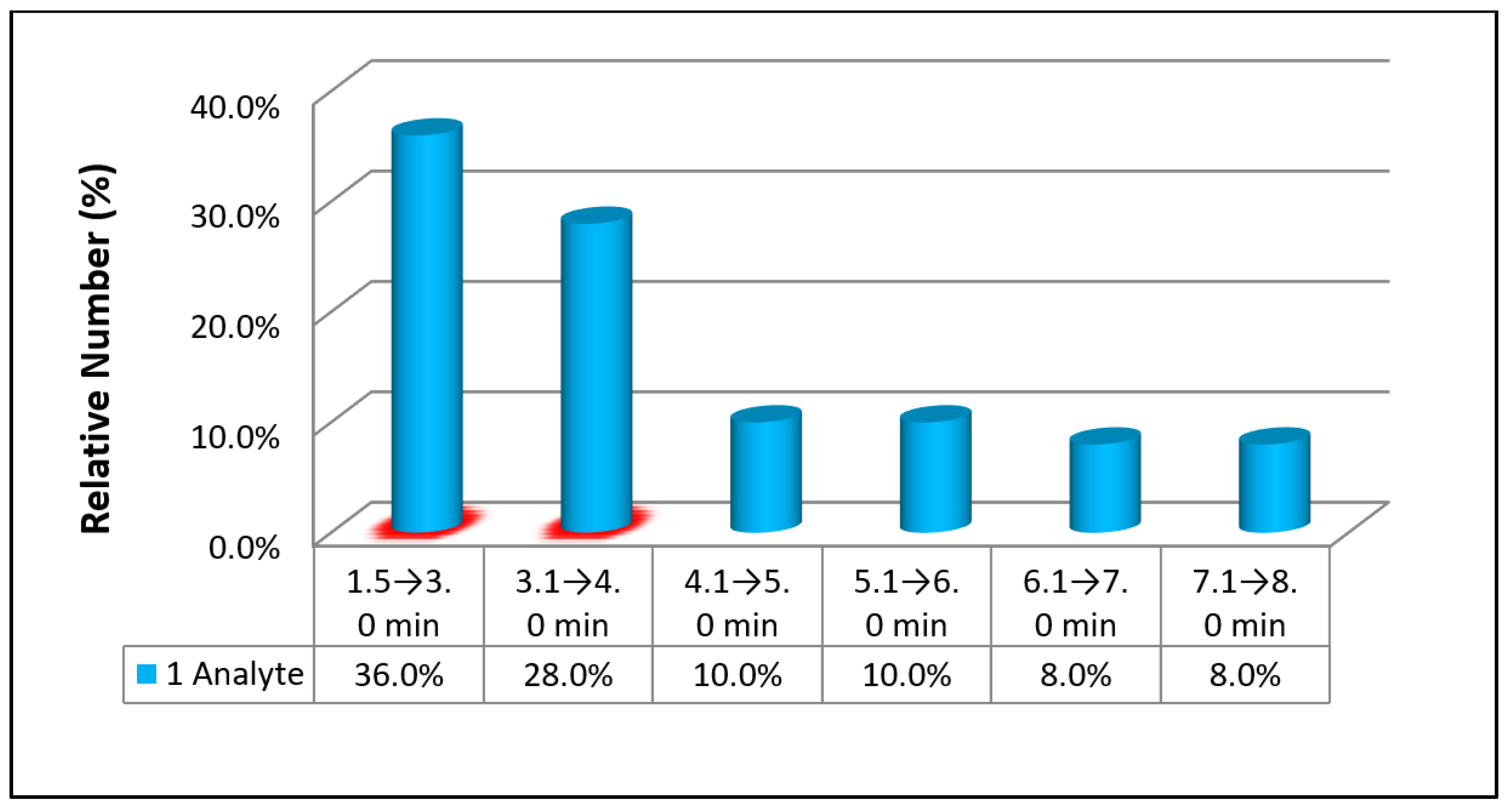
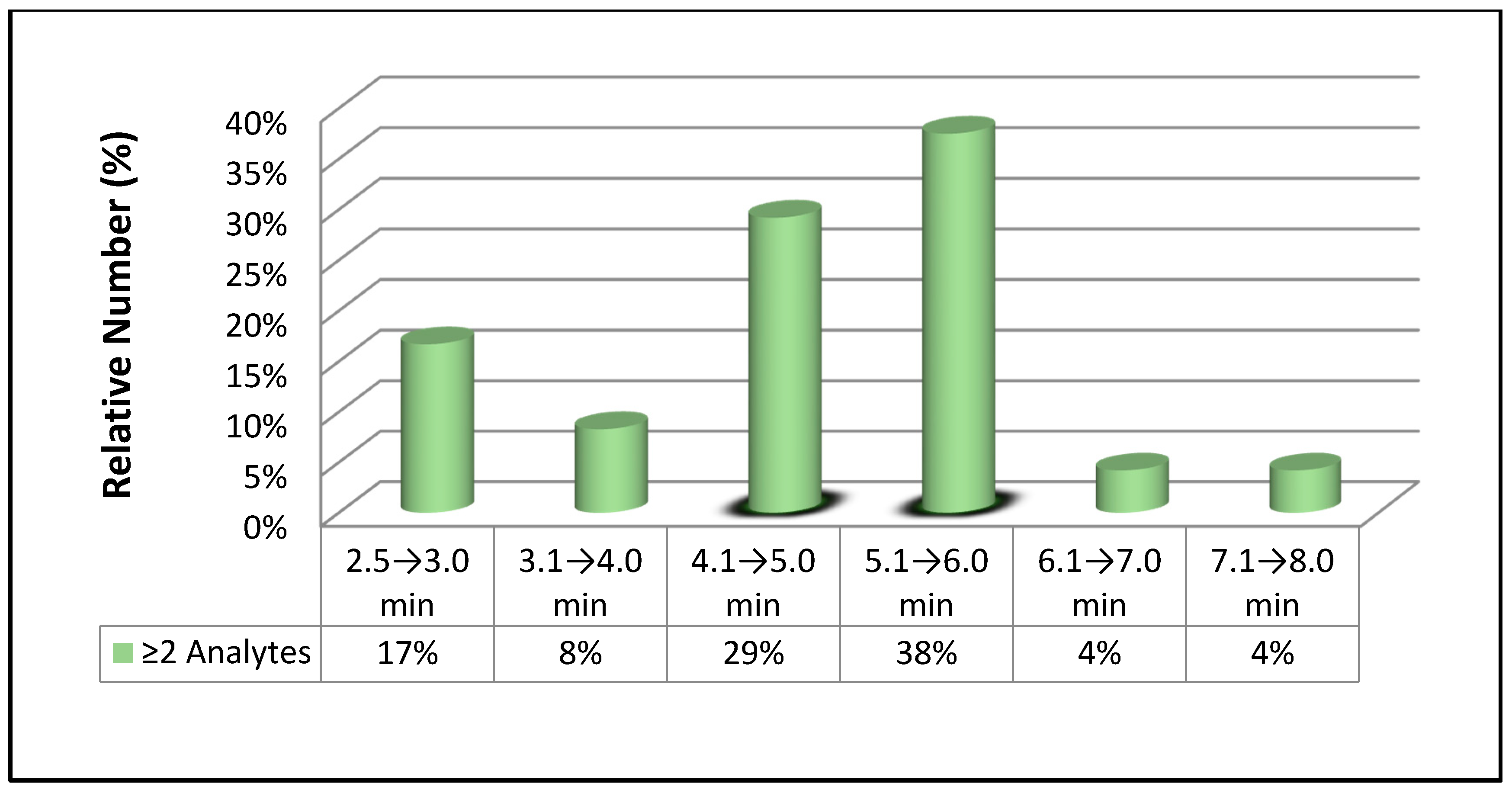
3.2. Chromatographic Separation
3.3. Matrix Effects
4. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Zhang, H.; Lin, Y.; Li, K.; Liang, J.; Xiao, X.; Cai, J.; Tan, Y.; Xing, F.; Mai, J.; Li, Y.; et al. Naturally Existing Oncolytic Virus M1 Is Nonpathogenic for the Nonhuman Primates After Multiple Rounds of Repeated Intravenous Injections. Hum. Gene Ther. 2016, 27, 700–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Dubey, R.; Roychowdhury, S.; Ghosh, M.; Sinha, B.N.; Kumar Pradhan, K.; Bal, T.; Muthukrishnan, V.; Seijas, J.A.; Pujarid, A. A rapid and sensitive determination of hypoxic radiosensitizer agent nimorazole in rat plasma by LC-MS/MS and its application to a pharmacokinetic study. Biomed. Chromatogr. 2015, 29, 1575–1580. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Dubey, N.K.; Dasgupta, A.K.; Sahu, M.; Benjamin, B.; Mullangi, R.; Srinivas, N.R. Highly sensitive method for the determination of JI-101, a multi-kinase inhibitor in human plasma and urine by LC-MS/MS-ESI: Method validation and application to a clinical pharmacokinetic study. Biomed. Chromatogr. 2012, 26, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Estella-Hermoso de Mendoza, A.; Imbuluzqueta, I.; Campanero, M.A.; Gonzalez, D.; Vilas-Zornoza, A.; Agirre, X.; Lana, H.; Abizanda, G.; Prosper, F.; Blanco-Prieto, M.J. Development and validation of ultra high performance liquid chromatography-mass spectrometry method for LBH589 in mouse plasma and tissues. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 3490–3496. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wang, Y.; Chang, J.; Zhang, J.; Wang, J.; Hu, X. Rapid and sensitive determination of vinorelbine in human plasma by liquid chromatography-tandem mass spectrometry and its pharmacokinetic application. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, N.V.; Koteshwara, M.; Vishwottam, K.N.; Puran, S.; Manoj, S.; Santosh, M. Simple, sensitive and rapid LC-MS/MS method for the quantitation of cerivastatin in human plasma—Application to pharmacokinetic studies. J. Pharm. Biomed. Anal. 2004, 36, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Deng, Z.; Sun, P.; Mu, Y.; Xue, M. Development and Validation of a Rapid and Sensitive LC-MS/MS Method for the Pharmacokinetic Study of Osimertinib in Rats. J. AOAC Int. 2017, 100, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Jangid, A.G.; Pudage, A.M.; Joshi, S.S.; Pabrekar, P.N.; Tale, R.H.; Vaidya, V.V. A simple, selective and rapid validated method for estimation of anastrazole in human plasma by liquid chromatography-tandem mass spectrometry and its application to bioequivalence study. Biomed. Chromatogr. 2010, 24, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Wang, X.D.; Zhong, G.P.; Qin, X.L.; Li, J.L.; Huang, Z.Y.; Zhu, X.; Li, M.F.; Huang, M. Development and validation of a highly rapid and sensitive LC-MS/MS method for determination of SZ-685C, an investigational marine anticancer agent, in rat plasma—Application to a pharmacokinetic study in rats. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Cannon, M.J.; Banach, M.; Pinchuk, A.N.; Ton, G.N.; Scheuerell, C.; Longino, M.A.; Weichert, J.P.; Tollefson, R.; Clarke, W.R.; et al. Quantification of CLR1401, a novel alkylphosphocholine anticancer agent, in rat plasma by hydrophilic interaction liquid chromatography-tandem mass spectrometric detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, S.; Zhao, M.; Mnatsakanyan, A.; Xu, L.; Ricklis, R.M.; Chen, A.; Karp, J.E.; Rudek, M.A. A rapid and sensitive method for determination of veliparib (ABT-888), in human plasma, bone marrow cells and supernatant by using LC/MS/MS. J. Pharm. Biomed. Anal. 2010, 52, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.Z.; Goh, B.C.; Grigg, M.E.; Lee, S.C.; Khoo, Y.M.; Lee, H.S. A rapid and sensitive liquid chromatography/tandem mass spectrometry method for determination of docetaxel in human plasma. Rapid Commun. Mass Spectrom. 2003, 17, 1548–1552. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Deng, Z.; Guo, H.; Ling, P. A rapid and sensitive determination of aucubin in rat plasma by liquid chromatography-tandem mass spectrometry and its pharmacokinetic application. Biomed. Chromatogr. 2012, 26, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Z.; Ong, R.Y.; Chin, T.M.; Thuya, W.L.; Wan, S.C.; Wong, A.L.; Chan, S.Y.; Ho, P.C.; Goh, B.C. Method development and validation for rapid quantification of hydroxychloroquine in human blood using liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2012, 61, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Minkin, P.; Zhao, M.; Chen, Z.; Ouwerkerk, J.; Gelderblom, H.; Baker, S.D. Quantification of sunitinib in human plasma by high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 874, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Peer, C.J.; Rao, M.; Spencer, S.D.; Shahbazi, S.; Steeg, P.S.; Schrump, D.S.; Figg, W.D. A rapid ultra HPLC-MS/MS method for the quantitation and pharmacokinetic analysis of 3-deazaneplanocin A in mice. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013, 927, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ahmad, A.; Ahmad, I. A sensitive and rapid liquid chromatography tandem mass spectrometry method for quantitative determination of 7-ethyl-10-hydroxycamptothecin (SN-38) in human plasma containing liposome-based SN-38 (LE-SN38). Biomed. Chromatogr. 2003, 17, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Corona, G.; Casetta, B.; Sandron, S.; Vaccher, E.; Toffoli, G. Rapid and sensitive analysis of vincristine in human plasma using on-line extraction combined with liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Rudek, M.A.; Mnasakanyan, A.; Hartke, C.; Pili, R.; Baker, S.D. A liquid chromatography/tandem mass spectrometry assay to quantitate MS-275 in human plasma. J. Pharm. Biomed. Anal. 2007, 43, 784–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, P.; Ko, Y.T. Validated LC-MS/MS method for simultaneous quantification of resveratrol levels in mouse plasma and brain and its application to pharmacokinetic and brain distribution studies. J. Pharm. Biomed. Anal. 2016, 119, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Johnson, J.; Wang, F.; Yang, L.; Broniscer, A.; Stewart, C.F. Determination of vandetanib in human plasma and cerebrospinal fluid by liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 2561–2566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, F.; Gu, Y.; Wang, T.; Gao, Y.; Li, X.; Gao, X.; Cheng, S. Quantification and pharmacokinetics of crizotinib in rats by liquid chromatography-tandem mass spectrometry. Biomed. Chromatogr. 2016, 30, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Li, J.; Ji, X.; Li, J.; Zhou, T.; Lu, W.; Li, L. An LC-MS/MS method for the quantitation of cabozantinib in rat plasma: Application to a pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 985, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Zhao, X.; Lu, X.; Wang, M.; Gong, M. HPLC-ESI-MS/MS validation and pharmacokinetics of kalopanaxsaponin A in rats. RSC Adv. 2015, 5, 7260–7266. [Google Scholar] [CrossRef]
- Tu, X.; Lu, Y.; Zhong, D.; Zhang, Y.; Chen, X. A sensitive LC-MS/MS method for quantifying clofarabine triphosphate concentrations in human peripheral blood mononuclear cells. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 964, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Albrecht, B.J.; Yan, X.; Gao, M.; Weng, H.R.; Bartlett, M.G. A rapid analytical method for the quantification of paclitaxel in rat plasma and brain tissue by high-performance liquid chromatography and tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2013, 27, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Ramagiri, S.; Ma, F.; Kosanam, H.; Wang, X.; Patil, R.; Miller, D.D.; Geisert, E.; Yates, C.R. Fast and sensitive liquid chromatography/electrospray mass spectrometry method to study ocular penetration of EDL-155, a novel antitumor agent for retinoblastoma in rats. J. Mass Spectrom. 2009, 44, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wang, Y.; Cao, J.; Li, J. Determination of henatinib in human plasma and urine by liquid chromatography-tandem mass spectrometry and its pharmacokinetic application. J. Pharm. Biomed. Anal. 2013, 80, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Heudi, O.; Vogel, D.; Lau, Y.Y.; Picard, F.; Kretz, O. Liquid chromatography tandem mass spectrometry method for the quantitative analysis of ceritinib in human plasma and its application to pharmacokinetic studies. Anal. Bioanal. Chem. 2014, 406, 7389–7396. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Sun, Q.; Liu, D.; Ma, B.; Zhao, H.; Fang, Z.; Wang, H.; Lou, H. A sensitive LC-MS/MS method to quantify methylergonovine in human plasma and its application to a pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1011, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Platova, A.I.; Miroshnichenko, I.I.; Ptitsina, S.N.; Yurchenko, N.I. Rapid and Sensitive LC-MS/MS Assay for Quantitation of Letrozole Using Solid-Phase Extraction from Human Blood Plasma and Its Application to Pharmacokinetic Studies. Pharm. Chem. J. 2014, 48, 292–297. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, J.; Xu, J.; Huang, Y.; Shen, Y.; Liu, Z. A rapid and sensitive LC-MS/MS assay for the quantitation of deacetyl mycoepoxydiene in rat plasma with application to preclinical pharmacokinetics studies. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 880, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jain, L.; Gardner, E.R.; Venitz, J.; Dahut, W.; Figg, W.D. Development of a rapid and sensitive LC-MS/MS assay for the determination of sorafenib in human plasma. J. Pharm. Biomed. Anal. 2008, 46, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, J.; Lu, T.; Zhang, J.; Wu, C.; Li, L.; He, Z.; Zhao, Y.; Liu, X. A rapid and sensitive LC-MS/MS method for evaluation of the absolute oral bioavailability of a novel c-Met tyrosine kinase inhibitor QBH-196 in rats. Biomed. Chromatogr. 2015, 29, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.I.; Nguyen, V.T.; Chen, M.L.; Adamson, P.C. A rapid, sensitive and selective liquid chromatography/atmospheric pressure chemical ionization tandem mass spectrometry method for determination of fenretinide (4-HPR) in plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 862, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernice, T.; Bishop, A.G.; Guillen, M.J.; Cuevas, C.; Aviles, P. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM01183 (lurbinectedin), a novel antineoplastic agent, in mouse, rat, dog, Cynomolgus monkey and mini-pig plasma. J. Pharm. Biomed. Anal. 2016, 123, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhong, B.; Su, B.; Xu, S.; Guo, B. Development and validation of a rapid LC-MS/MS method for the determination of JCC76, a novel antitumor agent for breast cancer, in rat plasma and its application to a pharmacokinetics study. Biomed. Chromatogr. 2012, 26, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Park, J.S.; Jo, M.H.; Park, M.S.; Ryu, J.H.; Cho, Y.W.; Shim, W.S.; Noh, G.J.; Lee, K.T. Rapid and sensitive LC-MS/MS method for determination of megestrol acetate in human plasma: Application to a human pharmacokinetic study. Biomed. Chromatogr. 2013, 27, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, J.; Yang, L.; Jia, Y.; Kong, L. A rapid and sensitive LC-MS/MS assay for the determination of berbamine in rat plasma with application to preclinical pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 929, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Wang, X.; Jia, Y.; Chu, Y.; Guan, X.; Ma, X.; Li, W.; Pan, G.; Zhou, S.; Sun, H.; et al. Quantitative determination of periplocymarin in rat plasma and tissue by LC-MS/MS: Application to pharmacokinetic and tissue distribution study. Biomed. Chromatogr. 2016, 30, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, W.; Yu, M.; Jiang, L. LC-MS/MS determination of 1-O-acetylbritannilactone in rat plasma and its application to a preclinical pharmacokinetic study. Biomed. Chromatogr. 2016, 30, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Bandu, R.; Ahn, H.S.; Lee, J.W.; Kim, Y.W.; Choi, S.H.; Kim, H.J.; Kim, K.P. Distribution study of cisplatin in rat kidney and liver cancer tissues by using liquid chromatography electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 2015, 50, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.M.; Jeong, Y.S.; Yim, C.S.; Lee, J.H.; Chung, S.J. Quantification of EC-18, a synthetic monoacetyldiglyceride (1-palmitoyl-2-linoleoyl-3-acetyl-rac-glycerol), in rat and mouse plasma by liquid-chromatography/tandem mass spectrometry. J. Pharm. Biomed. Anal. 2017, 137, 155–162. [Google Scholar] [CrossRef] [PubMed]
- De Krou, S.; Rosing, H.; Nuijen, B.; Schellens, J.H.; Beijnen, J.H. Fast and Adequate Liquid Chromatography-Tandem Mass Spectrometric Determination of Z-endoxifen Serum Levels for Therapeutic Drug Monitoring. Ther. Drug Monit. 2017, 39, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Anders, N.M.; Wanjiku, T.M.; He, P.; Azad, N.S.; Rudek, M.A. A robust and rapid liquid chromatography tandem mass spectrometric method for the quantitative analysis of 5-azacytidine. Biomed. Chromatogr. 2016, 30, 494–496. [Google Scholar] [CrossRef] [PubMed]
- De Bruijn, P.; Moghaddam-Helmantel, I.M.; de Jonge, M.J.; Meyer, T.; Lam, M.H.; Verweij, J.; Wiemer, E.A.; Loos, W.J. Validated bioanalytical method for the quantification of RGB-286638, a novel multi-targeted protein kinase inhibitor, in human plasma and urine by liquid chromatography/tandem triple-quadrupole mass spectrometry. J. Pharm. Biomed. Anal. 2009, 50, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Coward, L.U.; Freeman, L.; Noker, P.E.; Beattie, C.W.; Jia, L. A novel and rapid LC/MS/MS assay for bioanalysis of Azurin p28 in serum and its pharmacokinetics in mice. J. Pharm. Biomed. Anal. 2010, 53, 991–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coward, L.; Gorman, G.; Noker, P.; Kerstner-Wood, C.; Pellecchia, M.; Reed, J.C.; Jia, L. Quantitative determination of apogossypol, a pro-apoptotic analog of gossypol, in mouse plasma using LC/MS/MS. J. Pharm. Biomed. Anal. 2006, 42, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Rodin, I.; Braun, A.; Stavrianidi, A.; Shpigun, O. A validated LC-MS/MS method for rapid determination of methotrexate in human saliva and its application to an excretion evaluation study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 937, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Voggu, R.R.; Alagandula, R.; Zhou, X.; Su, B.; Zhong, B.; Guo, B. A rapid LC-MS/MS method for quantification of CSUOH0901, a novel antitumor agent, in rat plasma. Biomed. Chromatogr. 2015, 29, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Giri, K.K.; Suresh, P.S.; Saim, S.M.; Zainuddin, M.; Bhamidipati, R.K.; Dewang, P.; Hallur, M.S.; Rajagopal, S.; Rajagopal, S.; Mullangi, R. Validation of an LC-MS/MS method for simultaneous detection of four HDAC inhibitors—Belinostat, panobinostat, rocilinostat and vorinostat in mouse plasma and its application to a mouse pharmacokinetic study. Biomed. Chromatogr. 2017, 31, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Hu, P.; Jiang, J.; Kong, F.; Luo, H.; Zhao, Q. An UPLC-MS/MS method to determine CT-707 and its two metabolites in plasma of ALK-positive advanced non-small cell lung cancer patients. J. Pharm. Biomed. Anal. 2018, 153, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Z.; Lim, M.Y.; Chin, T.M.; Thuya, W.L.; Nye, P.L.; Wong, A.; Chan, S.Y.; Goh, B.C.; Ho, P.C. Rapid determination of gefitinib and its main metabolite, O-desmethyl gefitinib in human plasma using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 2155–2161. [Google Scholar] [CrossRef] [PubMed]
- Bouchet, S.; Chauzit, E.; Ducint, D.; Castaing, N.; Canal-Raffin, M.; Moore, N.; Titier, K.; Molimard, M. Simultaneous determination of nine tyrosine kinase inhibitors by 96-well solid-phase extraction and ultra performance LC/MS-MS. Clin. Chim. Acta 2011, 412, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Wang, X.; Liu, L.; Belinsky, M.G.; Kruh, G.D.; Gallo, J.M. Determination of methotrexate and its major metabolite 7-hydroxymethotrexate in mouse plasma and brain tissue by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2007, 43, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Merienne, C.; Rousset, M.; Ducint, D.; Castaing, N.; Titier, K.; Molimard, M.; Bouchet, S. High throughput routine determination of 17 tyrosine kinase inhibitors by LC-MS/MS. J. Pharm. Biomed. Anal. 2018, 150, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Shao, N.; Jin, Y.; Zhang, L.; Jiang, H.; Xiong, N.; Su, F.; Xu, H. Determination of non-liposomal and liposomal doxorubicin in plasma by LC-MS/MS coupled with an effective solid phase extraction: In comparison with ultrafiltration technique and application to a pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1072, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Rousset, M.; Titier, K.; Bouchet, S.; Dutriaux, C.; Pham-Ledard, A.; Prey, S.; Canal-Raffin, M.; Molimard, M. An UPLC-MS/MS method for the quantification of BRAF inhibitors (vemurafenib, dabrafenib) and MEK inhibitors (cobimetinib, trametinib, binimetinib) in human plasma. Application to treated melanoma patients. Clin. Chim. Acta 2017, 470, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.; Zeng, T.; Gao, S.; Xia, T.; Huang, L.; Zhang, F.; Chen, W. LC-MS/MS method for simultaneous determination of thalidomide, lenalidomide, cyclophosphamide, bortezomib, dexamethasone and adriamycin in serum of multiple myeloma patients. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1028, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Jiamboonsri, P.; Pithayanukul, P.; Bavovada, R.; Gao, S.; Hu, M. A validated liquid chromatography-tandem mass spectrometry method for the determination of methyl gallate and pentagalloyl glucopyranose: Application to pharmacokinetic studies. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 986–987, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Gao, Z.; Chen, X.; Zhong, D. Development and validation of a sensitive LC-MS/MS assay for the simultaneous quantification of allitinib and its two metabolites in human plasma. J. Pharm. Biomed. Anal. 2013, 86, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Kita, Y.; Iihara, H.; Yanase, K.; Ohno, Y.; Hirose, C.; Yamada, M.; Todoroki, K.; Kitaichi, K.; Minatoguchi, S.; et al. Simultaneous and rapid determination of gefitinib, erlotinib and afatinib plasma levels using liquid chromatography/tandem mass spectrometry in patients with non-small-cell lung cancer. Biomed. Chromatogr. 2016, 30, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Ekhart, C.; Gebretensae, A.; Rosing, H.; Rodenhuis, S.; Beijnen, J.H.; Huitema, A.D. Simultaneous quantification of cyclophosphamide and its active metabolite 4-hydroxycyclophosphamide in human plasma by high-performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry (LC-MS/MS). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 54, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Büttner, B.; Oertel, R.; Schetelig, J.; Middeke, J.M.; Bornhäuser, M.; Seeling, A.; Knoth, H. Simultaneous determination of clofarabine and cytarabine in human plasma by LC-MS/MS. J. Pharm. Biomed. Anal. 2016, 125, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Zhao, C.; He, X.R.; Jiang, S.T.; Han, S.Y.; Xu, G.B.; Li, P.P. Simultaneous determination of gefitinib and its major metabolites in mouse plasma by HPLC-MS/MS and its application to a pharmacokinetics study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1011, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Z.; Goh, S.H.; Wong, A.L.; Thuya, W.L.; Lau, J.Y.; Wan, S.C.; Lee, S.C.; Ho, P.C.; Goh, B.C. Validation of a rapid and sensitive LC-MS/MS method for determination of exemestane and its metabolites, 17β-hydroxyexemestane and 17β-hydroxyexemestane-17-O-β-D-glucuronide: Application to human pharmacokinetics study. PLoS ONE 2015, 10, e0118553. [Google Scholar] [CrossRef] [PubMed]
- Corona, G.; Elia, C.; Casetta, B.; Toffoli, G. Fast liquid chromatography-tandem mass spectrometry method for routine assessment of irinotecan metabolic phenotype. Ther. Drug Monit. 2010, 32, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhan, Y.; Chen, X.; Zhang, Y.; Zhong, D. Development and validation of a sensitive LC-MS/MS method for simultaneous quantification of sinotecan and its active metabolite in human blood. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 951–952, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Precht, J.C.; Ganchev, B.; Heinkele, G.; Brauch, H.; Schwab, M.; Mürdter, T.E. Simultaneous quantitative analysis of letrozole, its carbinol metabolite, and carbinol glucuronide in human plasma by LC-MS/MS. Anal. Bioanal. Chem. 2012, 403, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Wilson, W.R. Rapid and sensitive ultra-high-pressure liquid chromatography-tandem mass spectrometry analysis of the novel anticancer agent PR-104 and its major metabolites in human plasma: Application to a pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3181–3186. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, Z.; Che, J.; Meng, Q.; Shan, C.; Hou, Y.; Liu, X.; Chai, Y.; Cheng, Y. Development of a rapid and sensitive LC-MS/MS assay for the determination of combretastatin A4 phosphate, combretastatin A4 and combretastatin A4 glucuronide in beagle dog plasma and its application to a pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3813–3821. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zhou, J.; Zhang, F.; Miao, H.; Yun, Y.; Feng, J.; Tao, X.; Chen, W. Rapid and sensitive liquid chromatography coupled with electrospray ionization tandem mass spectrometry method for the analysis of paclitaxel, docetaxel, vinblastine, and vinorelbine in human plasma. Ther. Drug Monit. 2014, 36, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.S.; Phelps, M.A.; Blum, K.A.; Blum, W.; Grever, M.R.; Farley, K.L.; Dalton, J.T. Development and validation of a rapid and sensitive high-performance liquid chromatography-mass spectroscopy assay for determination of 17-(allylamino)-17-demethoxygeldanamycin and 17-(amino)-17-demethoxygeldanamycin in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 871, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Gumustas, M.; Kurbanoglu, S.; Uslu, B.; Ozkan, S.A. UPLC versus HPLC on drug analysis: Advantageous, applications and their validation parameters. Chromatographia 2013, 76, 1365–1427. [Google Scholar] [CrossRef]
- Cole, L.A.; Dorsey, J.G. Reduction of reequilibration time following gradient elution reversed-phase liquid chromatography. Anal. Chem. 1990, 62, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Farrell, J.T.; Lynn, K.; Euler, D.; Kwei, G.; Hwang, T.L.; Qin, X.Z. The importance of chromatographic separation in LC/MS/MS quantitation of drugs in biological fluids: Detection, characterization, and synthesis of a previously unknown low-level nitrone metabolite of a substance P antagonist. Anal. Chem. 2003, 75, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jia, J.; Aubry, A.; Arnold, M.; Jemal, M. Theory-guided efficient strategy to maximize speed and resolution in rapid gradient LC-MS/MS bioanalysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, G.; Tiwari, R. Bioanalytical method validation: An updated review. Pharm. Methods 2010, 1, 25–38. [Google Scholar] [CrossRef]
- King, R.; Bonfiglio, R.; Fernandez-Metzler, C.; Miller-Stein, C.; Olah, T. Mechanistic investigation of ionization suppression in electrospray ionization. J. Am. Soc. Mass Spectrom. 2000, 11, 942–950. [Google Scholar] [CrossRef]
- Taylor, P.J. Matrix effects: The Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin. Biochem. 2005, 38, 328–334. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte(s) | Indication | Matrices | Prep | Solid Phase | E-M | Interf | IS | ME (%) | RT (min) | LLOQ | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nimorazole | Radiosensitizer | r-plasma | PPT | C18 (50 × 4.6 mm, 2.7 µm) | ISO | ESI(+) | AN | NEG | 1.5 | 0.25 ng/mL | [4] |
| JI-101 | Multi-kinase inhibitor | h-plasma h-urine | SPE | C18 (50 × 2.1 mm, 5 µm) | ISO | ESI(+) | AN | 98 | 2.0 | 1.57 ng/mL 0.97 ng/mL | [5] |
| LBH589 | HDAC inhibitor | m-plasma m-tissue | LLE | C18 (50 × 2.1 mm, 1.7 µm) UPLC | ISO | ESI(+) | AN | NEG | 2.0 | 2.5 ng/mL 35.7 ng/mg | [6] |
| Vinorelbine | Vinca alkaloid | h-plasma | LLE | C18 (50 × 2.1 mm, 3 µm) | ISO | ESI(+) | AN | 95.8–106.7 | 2.0 | 0.1 ng/mL | [7] |
| Cerivastatin | Inhibitor of HMG-CoA reductase | h-plasma | LLE | C18 (100 × 3 mm, 3.5 µm) | ISO | ESI(+) | AN | NEG | 2.0 | 0.01 ng/mL | [8] |
| Osimertinib | Tyrosine kinase inhibitor | r-plasma | PPT | C18 (50 × 2.1 mm, 3 µm) | GRA | ESI(+) | AN | 90.1–97.3 | 2.5 | 1 ng/mL | [9] |
| Anastrazole | Aromatase inhibitor | h-plasma | SPE | C18 (50 × 4.6 mm, 5 μm) UPLC | ISO | ESI(+) | AN | 97.5 | 2.5 | 0.3 ng/mL | [10] |
| SZ-685C | Marine anticancer agent | r-plasma | LLE | C18 (100 × 2.1 mm, 3 µm) | ISO | ESI(−) | AN | 94.3 | 2.5 | 5 ng/mL | [11] |
| CLR1401 | Anticancer candidate | r-plasma | LLE | C18 (50 × 3.0 mm, 5 µm) | GRA | ESI(+) | IL | 80.0–86.0 | 2.8 | 2 ng/mL | [12] |
| Veliparib (ABT-888) | PARP-1 & 2 inhibitor | h-plasma | PPT | C18 (100 × 2.1 mm, 3 µm) | ISO | ESI(+) | AN | UNK | 3.0 | 5 nmol/L | [13] |
| Docetaxel | Anticancer drug | h-plasma | LLE | C8 (50 × 2.1 mm, 5 µm) | ISO | ESI(+) | AN | UNK | 3.0 | 5 ng/mL | [14] |
| Aucubin | Natural compound | r-plasma | PPT | Diamonsil C18(2) | ISO | ESI(+) | AN | 90.8–91.0 | 3.0 | 10 ng/mL | [15] |
| HCQ | Inhibitor of autophagy | h-blood | PPT | C8 (50 × 2.1 mm, 5 µm) | ISO | ESI(+) | IL | 93.0–100.6 | 3.0 | 5 ng/mL | [16] |
| Sunitinib | Tyrosine kinase inhibitor | h-plasma | LLE | C18 (50 × 2.1 mm, 3.5 µm) | ISO | ESI(+) | AN | UNK | 3.0 | 0.2 ng/mL | [17] |
| DZNep | Methylation inhibitor | m-plasma | LLE | HILIC (100 × 2.1 mm, 1.7 µm) UPLC | GRA | ESI(+) | AN | 84–87 | 3.0 | 5 ng/mL | [18] |
| SN-38 | Anticancer drug | h-plasma | PPT | C18 (50 × 2.0 mm, 4 µm) | GRA | ESI(+) | AN | UNK | 3.0 | 0.05 ng/mL | [19] |
| Vincristine | Anticancer drug | h-plasma | PPT | C18 (50 × 2.1 mm, 5 µm) | ISO | APCI(+) | AN | UNK | 3.0 | 0.1 ng/mL | [20] |
| MS-275 | HDAC inhibitor | h-plasma | LLE | C18(50 × 2.1 mm, 3.5 μm) | GRA | ESI(+) | AN | UNK | 3.0 | 0.5 ng/mL | [21] |
| trans-resveratrol | Natural compound | m-plasma m-brain | LLE | C18 (100 × 1 mm, 5 μm) | ISO | ESI(−) | AN | 93.8–100.6 | 3.0 | 5 ng/mL | [22] |
| ZD6474 | Tyrosine kinase inhibitor | h-plasma h-fluid | LLE | C18 (50 × 2.1 mm, 2.6 µm) | ISO | ESI(+) | IL | 98.0 | 3.0 | 0.25 ng/mL 0.25 ng/mL | [23] |
| Crizotinib | Tyrosine kinase inhibitor | r-plasma | PPT | Zorbax XDB C18 (2.1 × 50 mm, 3.5 μm) | GRA | ESI(+) | AN | 94.3–96.2 | 3.5 | 1 ng/mL | [24] |
| cabozantinib | Tyrosine kinase inhibitor | r-plasma | LLE | C18 (50 × 2 mm, 5 μm) | ISO | ESI(+) | AN | 105–115 | 3.5 | 0.5 ng/mL | [25] |
| KPS-A | Natural compound | r-plasma | PPT | C18 (2.1 × 50 mm, 3.5 μm) | GRA | ESI(+) | AN | 93–96 | 3.5 | 0.5 ng/mL | [26] |
| Clofarabine triphosphate | Metabolite of clofarabine | h-PBMC | PPT | CN (100 × 4.6 mm, 5 μm) | GRA | ESI(+) | AN | 91–105 | 3.5 | 1.25 ng/107 cells | [27] |
| Paclitaxel | Antimicrotubule agent | r-plasma r-tissue | LLE | C8 (50 × 2.1 mm, 5 μm) | ISO | ESI(+) | IL | 70.9–82.7 | 3.5 | 0.5 ng/mL 1.5 ng/g | [28] |
| EDL-155 | Anticancer agent | r-plasma | PPT | C8 (50 × 2.1 mm, 3.5 μm) | GRA | ESI(+) | AN | 98.6 | 3.5 | 0.1 ng/mL | [29] |
| Henatinib | Kinase inhibitor | h-plasma h-urine | PPT | C18 (50 × 2.1 mm, 2.5 μm) | ISO | ESI(+) | AN | 90.5–100.9 | 3.5 | 0.1 ng/mL 1 ng/mL | [30] |
| Ceritinib | ALK inhibitor | h-plasma h-brain | PPT | C18 (50 × 2.1 mm, 2.7 μm) | GRA | ESI(+) | IL | 92–109 | 3.6 | 1 ng/mL | [31] |
| Methergine | chemosensitizer for cancer | h-plasma | LLE | C18 (100 × 2.1 mm, 2.7 μm) | ISO | ESI(+) | AN | 61–66 | 4.0 | 0.025 ng/mL | [32] |
| Letrozole | Aromatase inhibitor | h-plasma | SPE | C18 (100 × 2.1 mm, 3.5 μm) | ISO | ESI(+) | AN | NEG | 4.0 | 0.25 ng/mL | [33] |
| Deacetyl mycoepoxydiene | Marine anticancer agent | r-plasma | PPT | C18 (150 × 2.1 mm, 5 μm) | ISO | ESI(+) | AN | 95.5–97.8 | 4.0 | 5 ng/mL | [34] |
| Sorafenib | Kinase inhibitor | h-plasma | PPT | SymmetryShield RP8 (50 × 2.1 mm, 3.5 μm) (0.1% FA:ACN) | ISO | ESI(+) | IL | 98.6 | 4.0 | 5 ng/mL | [35] |
| QBH-196 | c-Met tyrosine kinase inhibitor | r-plasma | LLE | C18 (50 × 2.1 mm, 2.6 µm) | GRA | ESI(+) | AN | 80–115 | 4.0 | 8 ng/mL | [36] |
| Fenretinide | Chemopreventive agent | m-plasma | PPT | C18 (50 × 2.1 mm, 5 μm) | GRA | APCI(+) | AN | 100.8–108.7 | 4.5 | 0.5 ng/mL | [37] |
| PM01183 | Antineoplastic agent | Animal plasma | SPE | C18 (30 × 2.1 mm, 3 μm) | GRA | ESI(+) | IL | 88–103 | 5.0 | 0.1 ng/mL | [38] |
| JCC76 | Antitumor agent | r-plasma | LLE | C18 (40 × 2.0 mm, 5 μm) | ISO | ESI(−) | AN | 90.8–96.9 | 5.0 | 0.3 ng/mL | [39] |
| Megestrol acetate | Hormonal therapy | h-plasma | LLE | C18 (50 × 2.0 mm, 3 µm) | ISO | ESI(+) | AN | 92.3–95.8 | 5.0 | 1.0 ng/mL | [40] |
| Berbamine | Natural compound | r-plasma | PPT | C18 (150 × 2.0 mm, 5 μm) | GRA | ESI(+) | AN | 97.2–98.5 | 5.5 | 1 ng/mL | [41] |
| Peri-plocymarin | potential anticancer agent | r-plasma r-tissue | LLE | C18 (2.1 × 150 mm, 3.0 μm) | ISO | ESI(+) | AN | 95.8–105 | 6.0 | 0.5 ng/mL | [42] |
| ABL | potential anticancer agent | r-plasma | PPT | C18 (50 × 4.6 mm, 3.0 μm) | ISO | ESI(+) | AN | 104–108 | 6.0 | 1.6 ng/mL | [43] |
| Cisplatin | Anticancer drug | r-tissue | LLE | C18 (50 × 2.1 mm, 1.8 μm) | ISO | ESI(+) | AN | 89–104 | 6.0 | 5 ng/mL | [44] |
| EC-18 | Anticancer agent | r-plasma m-plasma | PPT | C18 (150 × 2 mm, 4.0 μm) | GRA | ESI(+) | IL | 77.9–89.0 | 7.0 | 50 ng/mL | [45] |
| Z-endoxifen | Anti-estrogen | h-serum | PPT | C18 (150 × 2.1 mm, 2.6 µm) | GRA | ESI(+) | IL | NA | 7.0 | 1 ng/mL | [46] |
| 5-azacytidine | Anticancer agent | h-plasma | SPE | C18 (250 × 2.1 mm, 4 µm) | ISO | ESI(+) | AN | 51–55 | 7.0 | 5 ng/mL | [47] |
| RGB-286638 | Protein kinase inhibitor | h-plasma h-urine | LLE | C18 (50 × 2.1 mm, 5 µm) | GRA | ESI(+) | IL | 146–151 | 7.0 | 2 ng/mL 2 ng/mL | [48] |
| Azurin p28 | Anticancer peptide | m-ser | PPT | C18 (100 × 2 mm, 5 µm) | GRA | ESI(+) | AN | UNK | 7.5 | 100 ng/mL | [49] |
| Apogossypol | Bcl-2 inhibitor | m-plasma | PPT | C18 (100 × 2 mm, 4 µm) | GRA | ESI(+) | AN | UNK | 7.5 | 10 ng/mL | [50] |
| Methotrexate | Anticancer drug | h-saliva | SPE | C18 (150 × 2.0 mm, 2.2 μm) | GRA | ESI(+) | AN | 96–104 | 8.0 | 1.0 ng/mL | [51] |
| CSUOH0901 | COX-2 inhibitor | r-plasma | PPT | C18 (50 × 2.0 mm, 5 μm) | GRA | ESI(+) | AN | 90.1–94.5 | 8.0 | 0.5 ng/mL | [52] |
| Analyte(s) | Indication | Matrices | Prep | S-Ph | E-Mode | Interf | IS | ME (%) | RT (min) | LLOQ | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Belinostat Panobinostat Rocilinostat Vorinostat | HDAC inhibitor | r-plasma | PPT | C18 (50 × 4.6 mm, 5 μm) | ISO | ESI(+) | AN | No significant ME | 2.5 | 2.9 ng/mL 2.9 ng/mL 1.0 ng/mL 1.0 ng/mL | [53] |
| CT-707 CT-707M1 CT-707M2 | Tyrosine kinase selective inhibitor | h-plasma | SPE | C18 (2.1 × 50 mm, 1.7 μm) UPLC | GRA | ESI(+) | IL | 86.9–102 | 3.0 | 2 ng/mL 1 ng/mL 1 mg/mL | [54] |
| Gefitinib O-DMG | EGFR inhibitor | h-plasma | PPT | C18 (150 × 2.1 mm, 5 µm) | ISO | ESI(+) | AN | 93.0–103.3; 41.6–50.2 | 3.0 | 5 nmol/L | [55] |
| Sunitinib Gefitinib Norimatinib (met) Imatinib Dasatinib Erlotinib Axitinib Nilotinib Lapatinib Sorafenib | Nine tyrosine kinase inhibitors and one metabolite of Imatinib | h-pls | SPE | C18 (50 × 2.1 mm, 1.7 μm) UPLC | GRA | ESI(+) | IL | 96.6 104.5 85.5 85.0 84.5 81.6 113.1 101.8 91.2 107.7 | 4.0 | 10 ng/mL 0.1 ng/mL 10 mg/mL 10 ng/mL 0.1 ng/mL 10 mg/mL 0.1 ng/mL 10 ng/mL 10 mg/mL 0.1 mg/mL | [56] |
| MTX 7-OH-MTX | Anticancer drug | m-plasma m-brain | PPT | C18 (50 × 2.0 mm, 5 µm) | ISO | ESI(+) | IL | 88.2–108.8 | 4.0 | 3.7 ng/mL 7.4 ng/mL | [57] |
| 17 tyrosine kinase inhibitors | EGFR tyrosine kinase inhibitors | h-plasma | SPE | C18 (5 × 2.1 mm, 1.6 µm) UPLC | GRA | ESI(+) | IL | 83.4–116.40 | 5.0 | 0.1 ng/mL | [58] |
| Doxorubicin L-DOX | Anticancer antibiotic | h-plasma | SPE | C18 (50 × 2.1 mm, 5.0 μm) | GRA | ESI(+) | AN IL | 95.7–98.9 | 5.0 | 3.13 ng/mL 0.15 μg/mL | [59] |
| Vemurafenib, Dabrafenib Cobimetinib, Trametinib Binimetinib | 2 BRAF inhibitors 3 MEK inhibitors | h-plasma | SPE | C18 (100 × 2.1 mm, 5.0 μm) UPLC | GRA | ESI(+) | IL | 80.6–115.4 | 5.0 | 0.4 ng/mL 1.0 ng/mL 0.5 ng/mL 0.5 ng/mL 0.75 ng/mL | [60] |
| Thalidomide Lenalidomide Cyclophosphamide Bortezomib Dexamethasone Adriamycin | Anticancer drug | h-serum | SPE | C18 (50 × 2.1 mm, 2.5 μm) | GRA | ESI(+) | AN | 89–100 60–64 113–124 103–126 90–92 143–163 | 5.0 | 4 ng/mL 2 ng/mL 2 ng/mL 2 ng/mL 2 ng/mL 2 ng/mL | [61] |
| MG PGG | Natural compounds | r-blood | LLE | C18 (50 × 2.1 mm, 5 µm) UPLC | GRA | ESI(+) | AN | 76–87 80–104 | 5.0 | 19.5 nmol/L 39 nmol/L | [62] |
| Allitinib M6 M10 | Irreversible inhibitor of the EGFR 1/2 | h-plasma | PPT | C18 (50 × 4.6 mm, 1.8 µm) | GRA | ESI(+) | AN | 98.7–105.0 | 5.0 | 0.3 ng/mL 0.03 ng/mL 0.075 ng/mL | [63] |
| Gefitinib Erlotinib Afatinib | EGFR tyrosine kinase inhibitors | h-plasma | LLE | C18 (50 × 2.1 mm, 3.5 µm) UPLC | ISO | ESI(+) | AN | UNK | 5.0 | 0.01 nmol/L 0.01 nmol/L 0.05 nmol/L | [64] |
| CP 4OHCP | Anticancer drug | h-plasma | PPT | C18 (150 × 2.1 mm, 5 μm) | GRA | ESI(+) | IL AN | UNK | 6.0 | 0.2 µg/mL 0.05 µg/mL | [65] |
| Clofarabine Cytarabine | Anticancer drug | h-plasma | PPT | C18 (150 × 2.0 mm, 4 μm) | GRA | ESI(+) | AN | None | 6.0 | 8 ng/mL 20 ng/mL | [66] |
| Gefitinib M523595 M537194 M387783 M608236 | EGFR tyrosine kinase inhibitor & its metabolites | m-plasma | PPT | C18 (50 × 2.1 mm, 1.8 m) | GRA | ESI(+) | AN | 86–112 | 6.0 | 1 ng/mL 1 ng/mL 1 ng/mL 1 ng/mL 1 ng/mL 0.5 ng/mL | [67] |
| Exemestane 17β-2H-EXE 17β-2H-EXE-Glu | Steroidal aromatase inhibitor | h-plasma | PPT | C18 (100 × 2.1 mm, 5 µm) | GRA | ESI(+) | AN | 62.2 54.2 33.8 | 6.0 | 0.4 ng/mL 0.2 ng/mL 0.2 ng/mL | [68] |
| CPT-11 SN-38 SN-38G APC NPC | Topoisomerase I inhibitor | h-plasma | PPT | C18 (50 × 2.0 mm, 2.6 µm) | GRA | ESI(+) | AN | 91.0 | 6.0 | 0.5 ng/mL 0.2 ng/mL 0.5 ng/mL 0.5 ng/mL 0.2 ng/mL | [69] |
| Sinotecan 7-HEC | Anticancer agent | h-blood | PPT | C8 (150 × 4.6 mm, 5 µm) | GRA | ESI(+) | AN | 104–114 | 6.0 | 1 ng/mL 0.5 ng/mL | [70] |
| Letrozole Carbinol carbinol glucuronide | Aromatase inhibitor and its metabolites | h-pls | SPE | C18 (50 × 4.6 mm, 1.8 μm) UPLC | GRA | ESI(+) | IL | 30–31 90–100 | 6.0 | 20 nmol/L 0.2 nmol/L 2 nmol/L | [71] |
| PR104 PR-104A PR-104G PR-104H PR-104M | Hypoxia-activated prodrug | h-plasma | PPT | C18 (50 × 2.1 mm, 1.8 µm) UPLC | GRA | ESI(+) | IL | 87.4–112.6 | 6.0 | 0.1 µmol/L 0.05 µmol/L 0.05 µmol/L 0.025 µmol/L 0.01 µmol/L | [72] |
| CA4P CA4 CA4-Glu | Antitumor vascular disrupting agent | d-plasma | PPT | C18 (150 × 3.0 mm, 5 μm) | GRA | ESI(+) ESI(+) ESI(−) | AN | NEG | 6.0 | 5 ng/mL | [73] |
| Paclitaxel Docetaxel Vinblastine Vinorelbine | Regulators of microtubule formation | h-plasma | LLE | C18 (100 × 2.1 mm, 3.5 µm) | ISO | ESI(+) | AN | 86.7–102.5 | 6.0 | 25 ng/mL 10 ng/mL 10 ng/mL 10 ng/mL | [74] |
| 17AAG 17AG | HSP90 inhibitor | h-plasma | PPT | C18 (50 × 2.1 mm, 5 μm) | GRA | ESI(+) | AN | UNK | 7.0 | 0.5 ng/mL 0.5 ng/mL | [75] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, A.L.-A.; Xiang, X.; Ong, P.S.; Mitchell, E.Q.Y.; Syn, N.; Wee, I.; Kumar, A.P.; Yong, W.P.; Sethi, G.; Goh, B.C.; et al. A Review on Liquid Chromatography-Tandem Mass Spectrometry Methods for Rapid Quantification of Oncology Drugs. Pharmaceutics 2018, 10, 221. https://doi.org/10.3390/pharmaceutics10040221
Wong AL-A, Xiang X, Ong PS, Mitchell EQY, Syn N, Wee I, Kumar AP, Yong WP, Sethi G, Goh BC, et al. A Review on Liquid Chromatography-Tandem Mass Spectrometry Methods for Rapid Quantification of Oncology Drugs. Pharmaceutics. 2018; 10(4):221. https://doi.org/10.3390/pharmaceutics10040221
Chicago/Turabian StyleWong, Andrea Li-Ann, Xiaoqiang Xiang, Pei Shi Ong, Ee Qin Ying Mitchell, Nicholas Syn, Ian Wee, Alan Prem Kumar, Wei Peng Yong, Gautam Sethi, Boon Cher Goh, and et al. 2018. "A Review on Liquid Chromatography-Tandem Mass Spectrometry Methods for Rapid Quantification of Oncology Drugs" Pharmaceutics 10, no. 4: 221. https://doi.org/10.3390/pharmaceutics10040221
APA StyleWong, A. L. -A., Xiang, X., Ong, P. S., Mitchell, E. Q. Y., Syn, N., Wee, I., Kumar, A. P., Yong, W. P., Sethi, G., Goh, B. C., Ho, P. C. -L., & Wang, L. (2018). A Review on Liquid Chromatography-Tandem Mass Spectrometry Methods for Rapid Quantification of Oncology Drugs. Pharmaceutics, 10(4), 221. https://doi.org/10.3390/pharmaceutics10040221