Novel Formulations of C-Peptide with Long-Acting Therapeutic Potential for Treatment of Diabetic Complications


 ,
, 

Abstract
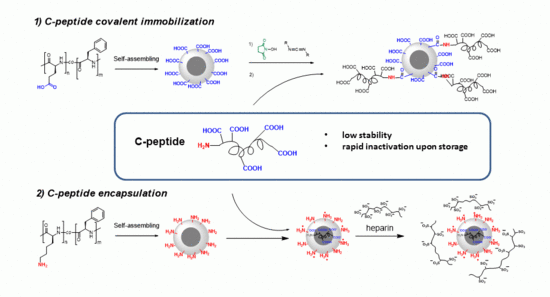
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
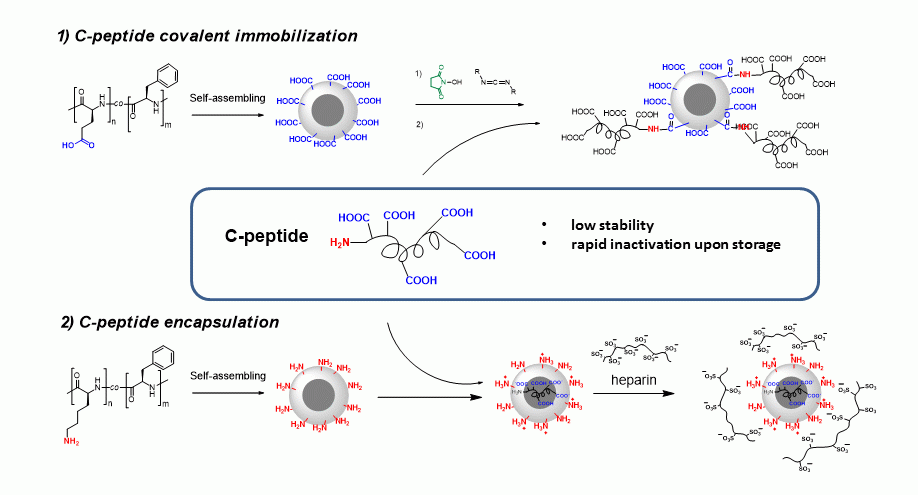
2.2.1. Synthesis and Polymer Characterization
2.2.2. Preparation and Characterization of Nanospheres
2.2.3. Synthesis of C-Terminal Fragment of C-Peptide
2.2.4. In Vitro Biodegradation Study
2.2.5. Particle Surface Modification with Peptides
2.2.6. Encapsulation of C-Peptide
2.2.7. Drug Release
2.2.8. Cell Culture Experiments
2.2.9. Microcalorimetric Ex Vivo Assay of Na+/K+-ATPase on Living Erythrocytes
3. Results and Discussion
3.1. Polymerization and Polymer Characterization
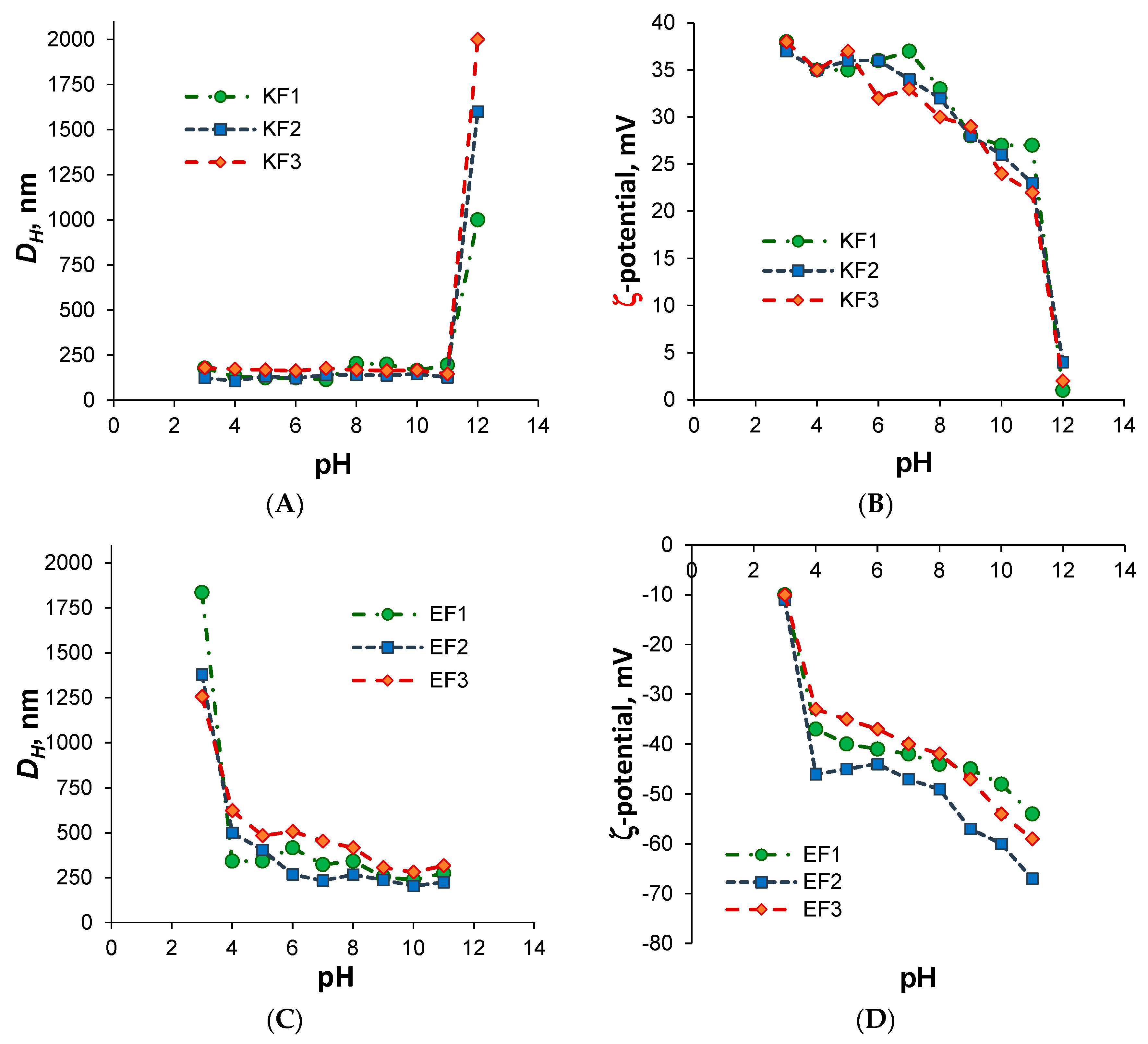
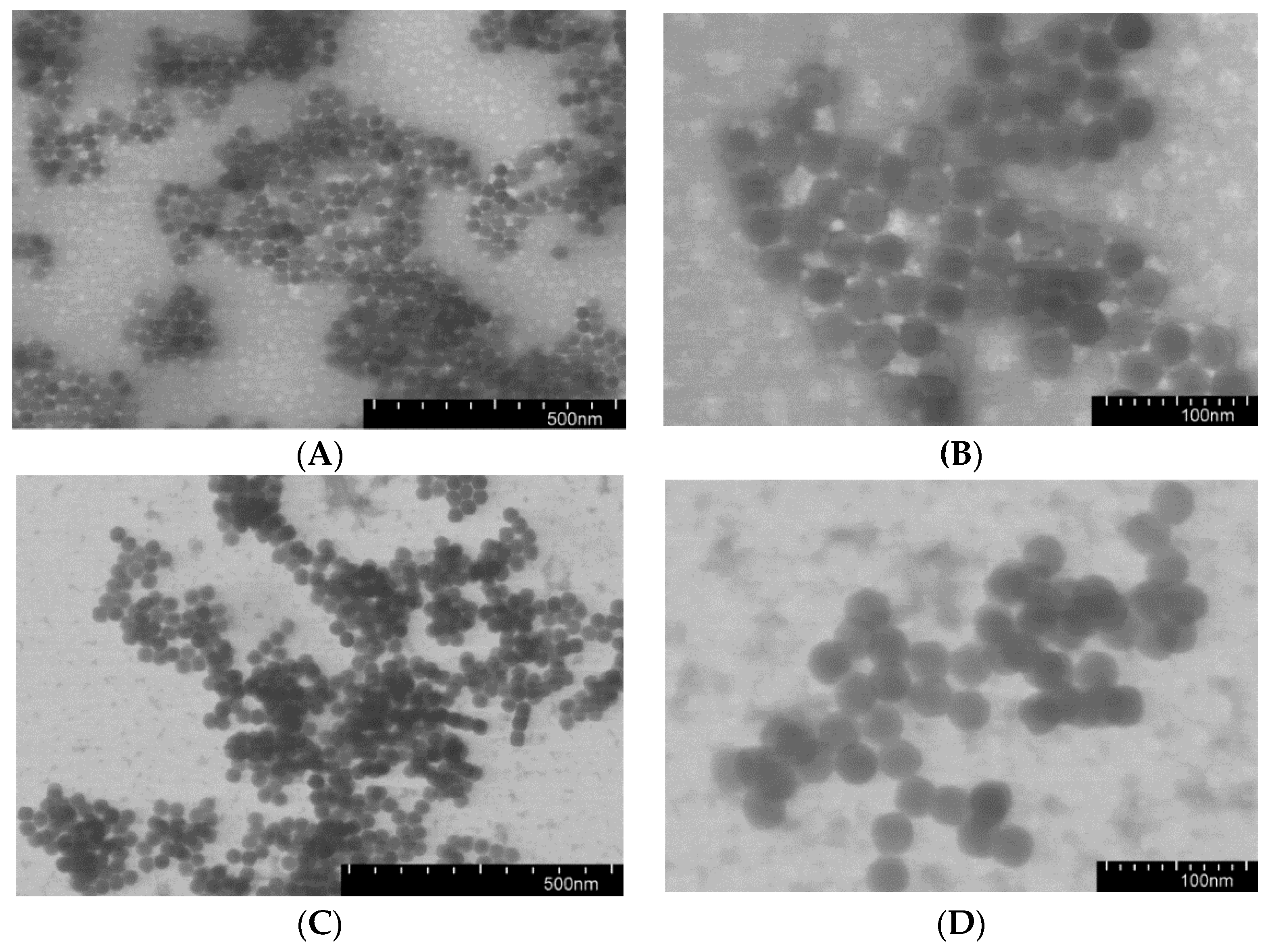
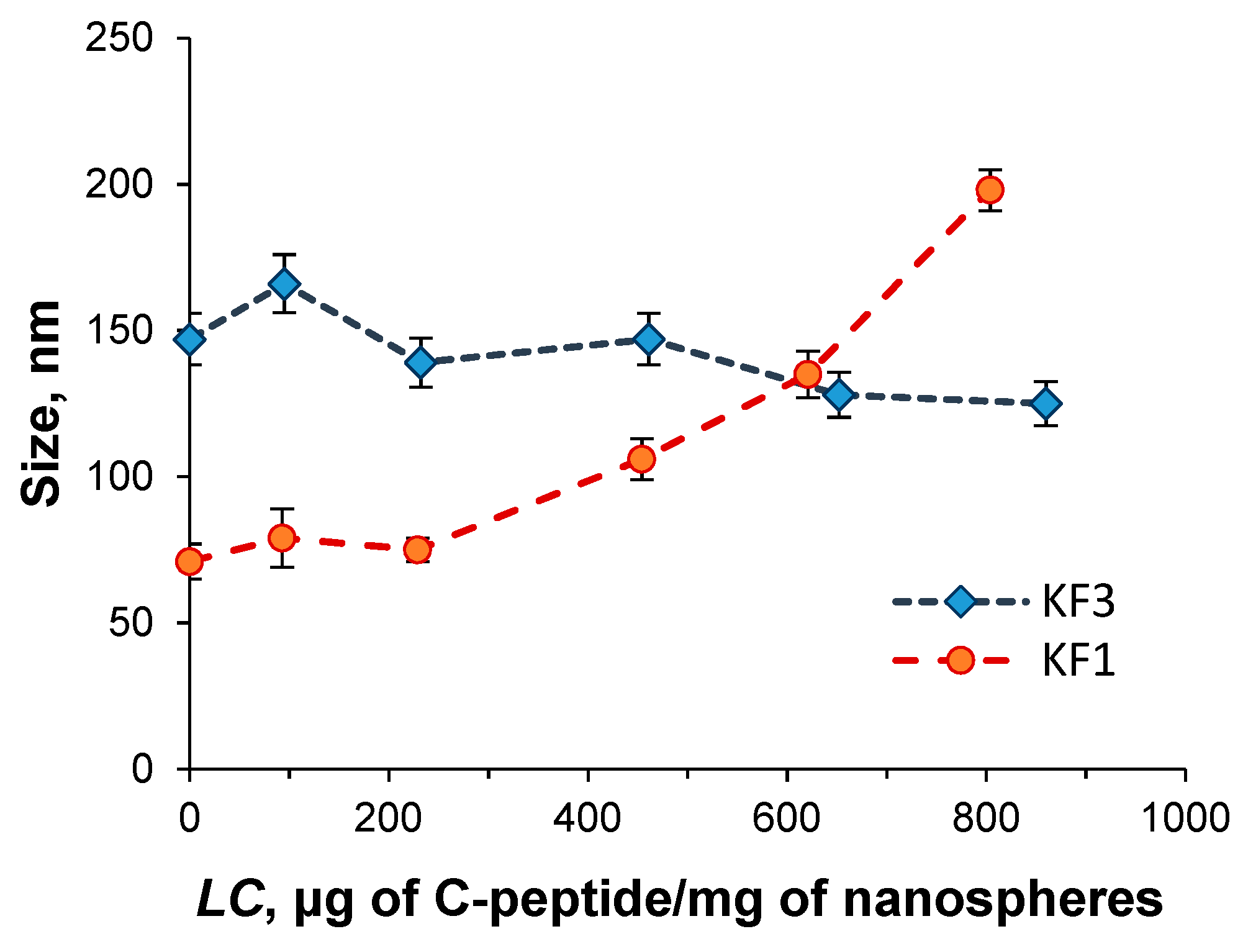
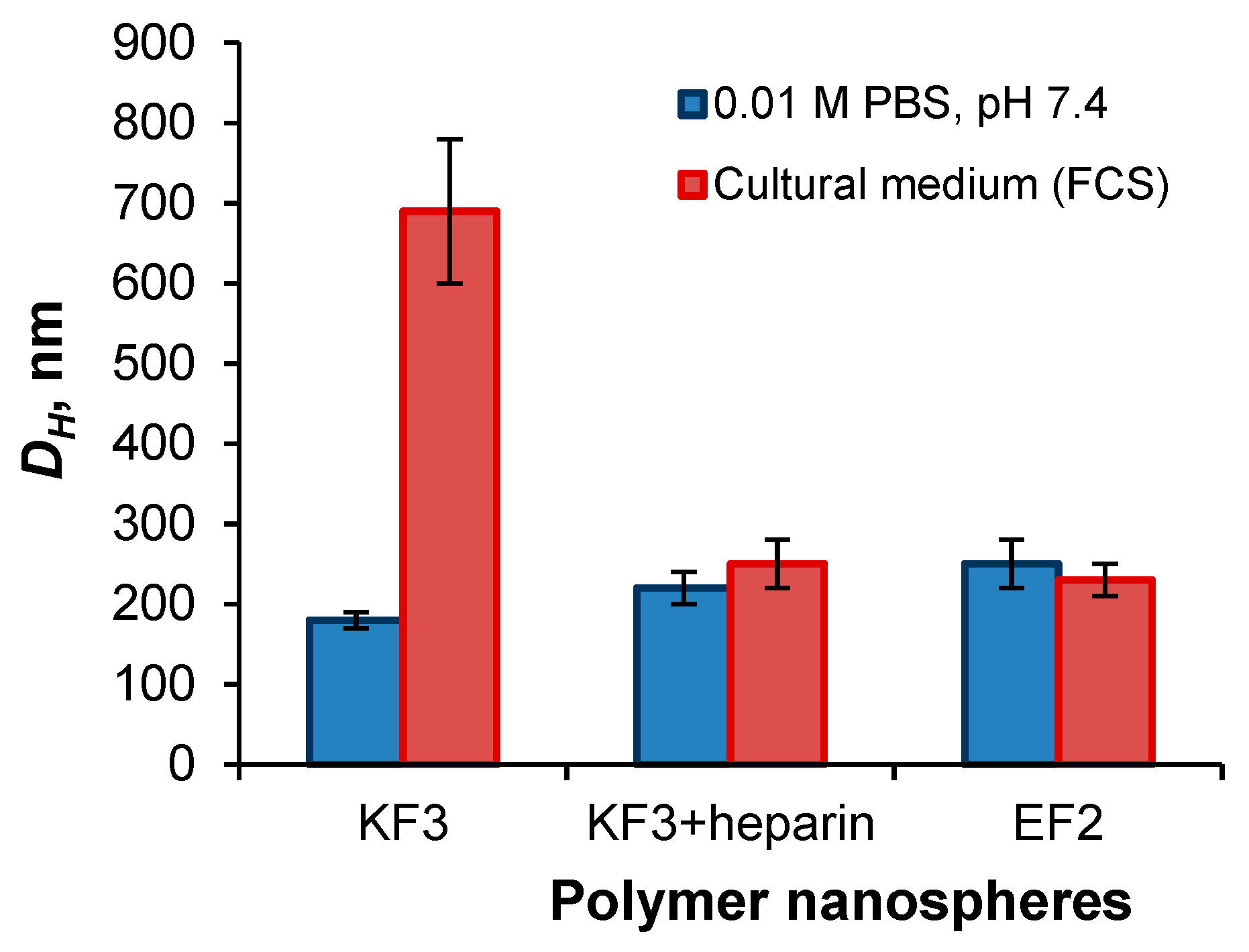
3.2. Preparation and Characterization of Nanospheres
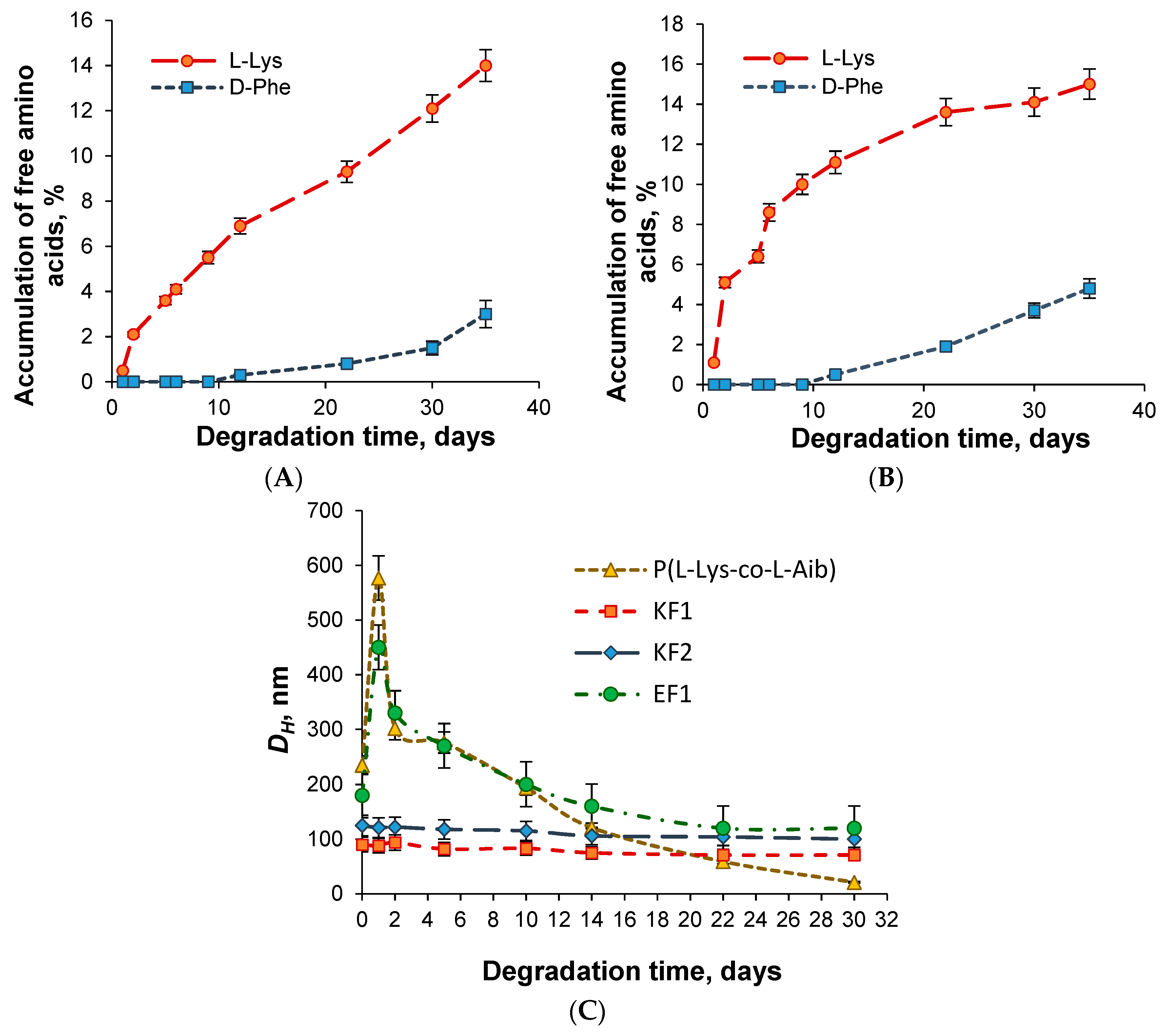
3.3. In Vitro Biodegradation Study
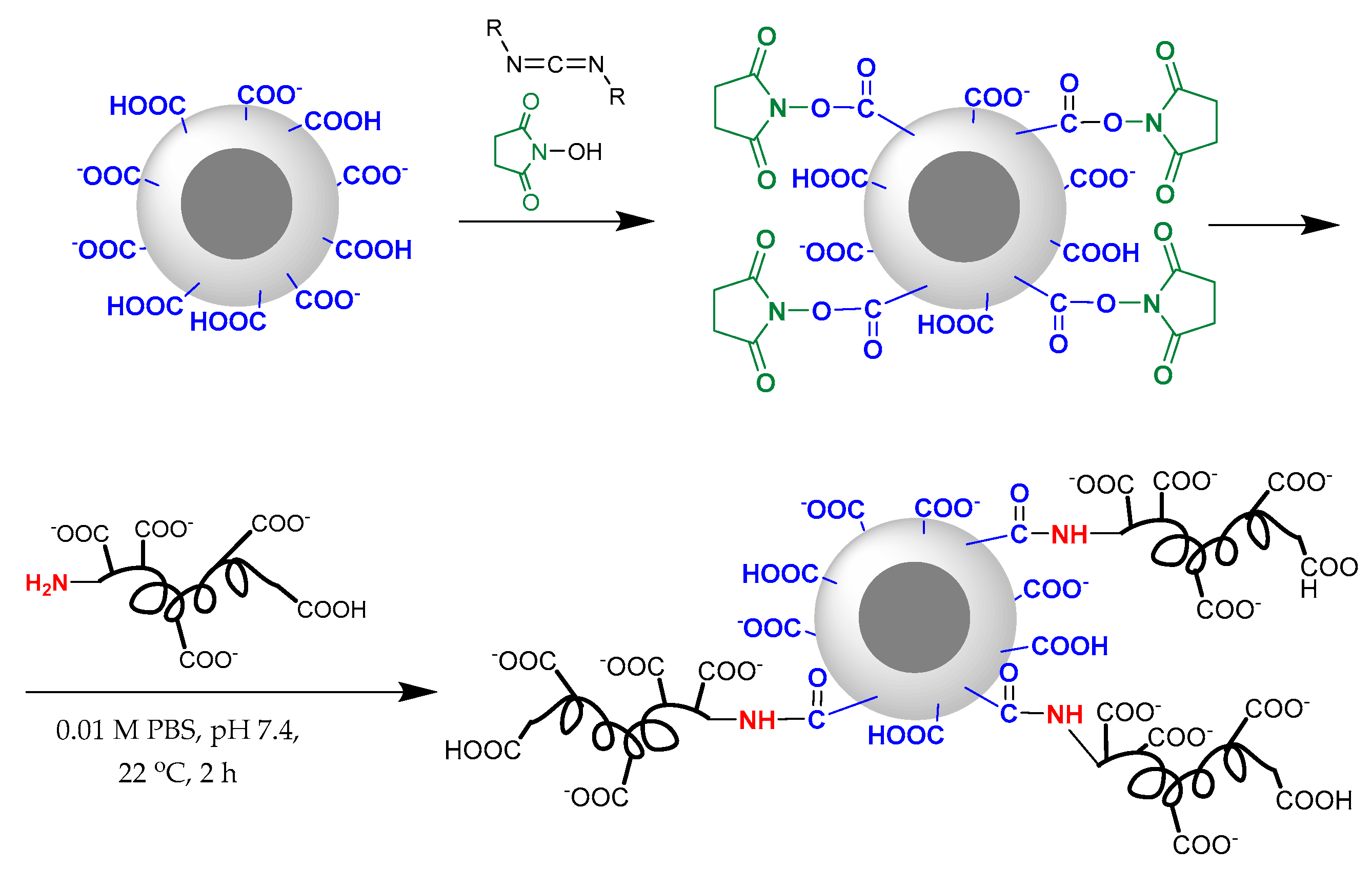
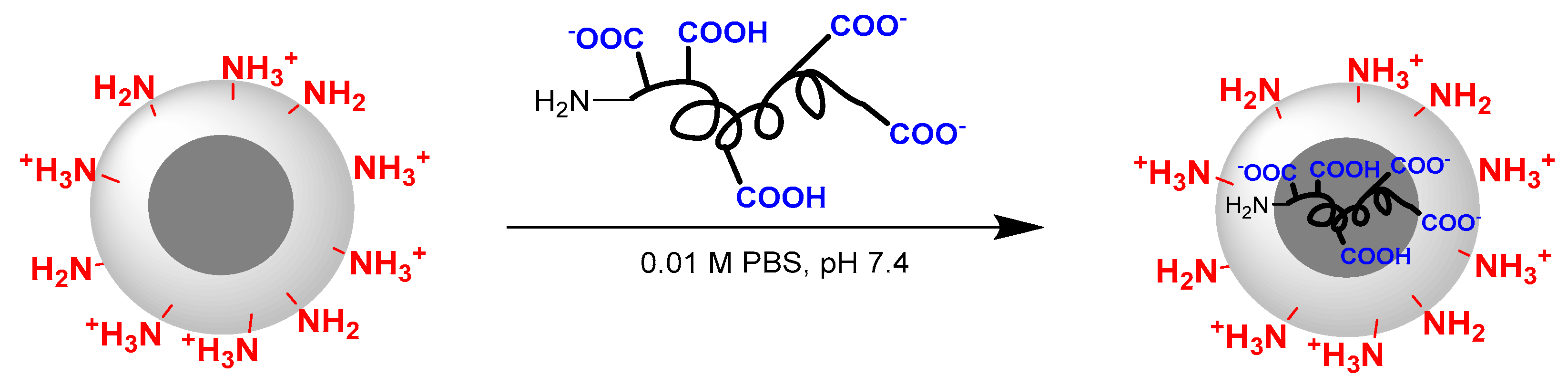
3.4. Surface Modification
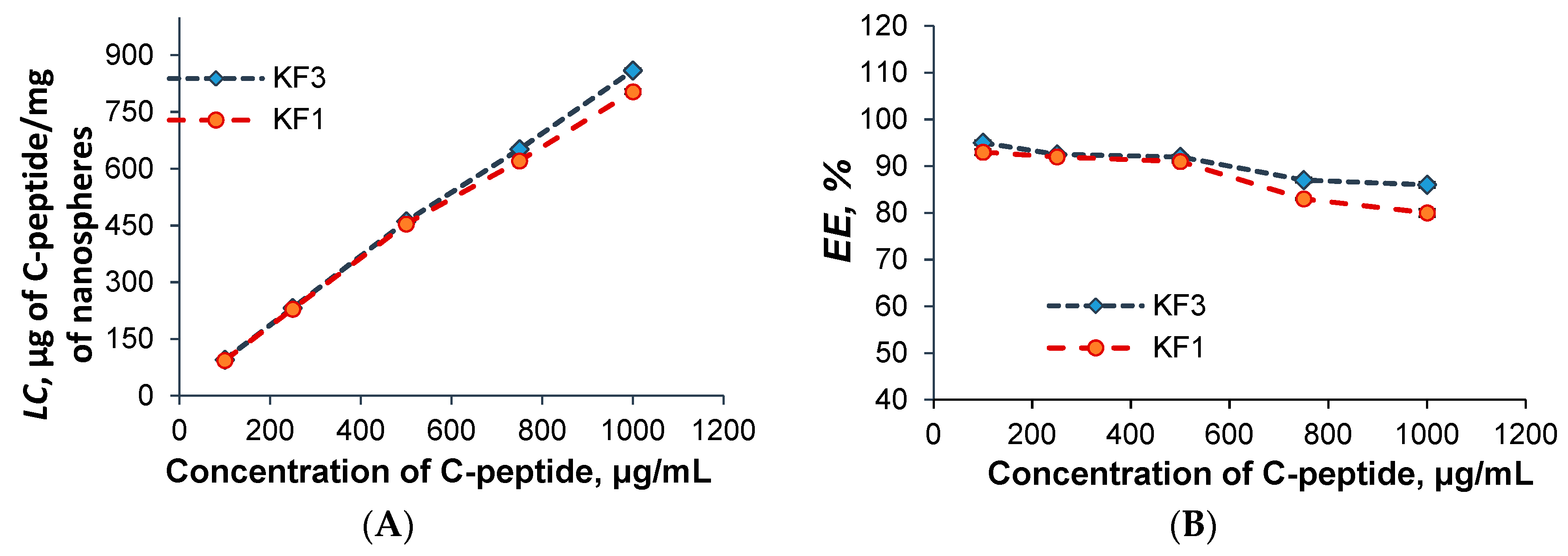
3.5. C-Peptide Encapsulation
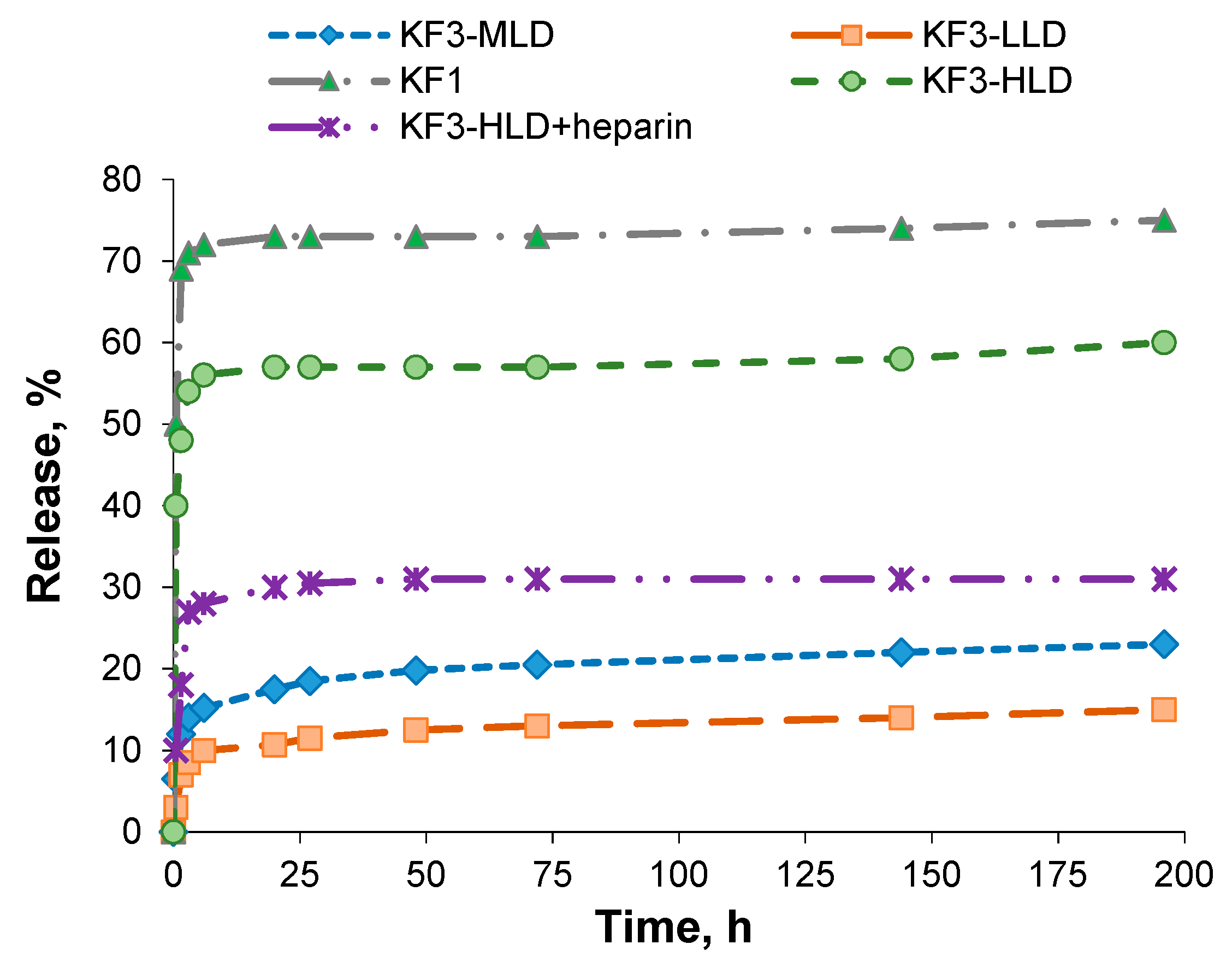
3.6. Drug Release
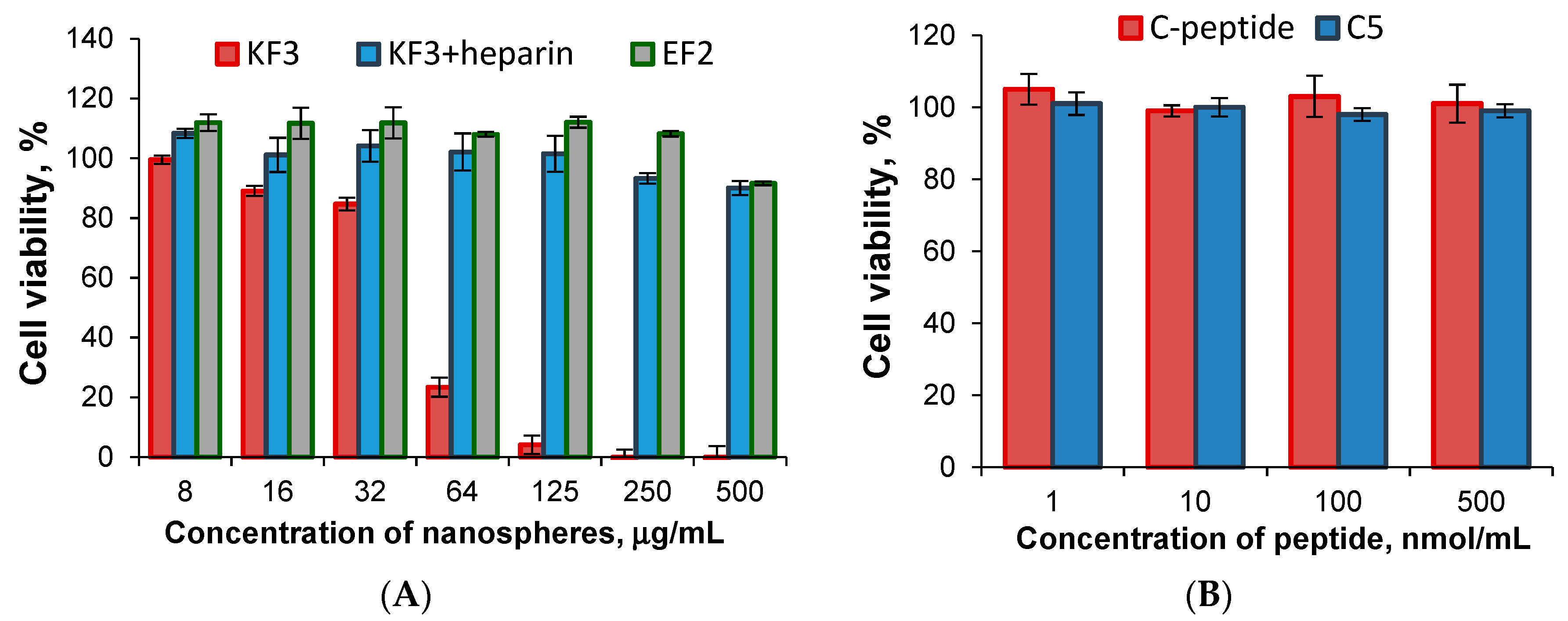
3.7. Cell Culture Experiments
3.8. Microcalorimetric Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sima, A.A.F. Diabetes & C-Peptide: Scientific and Clinical Aspects; Springer: New York, NY, USA, 2012; pp. 1–169. [Google Scholar]
- Luzi, L.; Zerbini, G.; Caumo, A. C-peptide: A redundant relative of insulin? Diabetologia 2007, 50, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, D.D.; Li, Y.; Meng, L.; Enwer, G. Serum C-peptide as a key contributor to lipid-related residual cardiovascular risk in the elderly. Arch. Gerontol. Geriatr. 2017, 73, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Shpakov, A.O. Mechanisms of Action and Therapeutic Potential of Proinsulin C-peptide. J. Evol. Biochem. Physiol. 2017, 53, 180–190. [Google Scholar] [CrossRef]
- Haidet, J.; Cifarelli, V.; Trucco, M.; Luppi, P. C-peptide reduces pro-inflammatory cytokine secretion in LPS-stimulated U937 monocytes in condition of hyperglycemia. Inflamm. Res. 2012, 61, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Leighton, E.; Sainsbury, C.A.; Jones, G.C. A Practical Review of C-Peptide Testing in Diabetes. Diabetes Ther. 2017, 8, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walcher, D.; Babiak, C.; Poletek, P.; Rosenkranz, S.; Bach, H.; Betz, S.; Durst, R.; Grub, M.; Hombach, V.; Strong, J.; et al. C-peptide induces vascular smooth muscle cell proliferation: Involvementof SRC-kinase, phosphatidylinositol 3-kinase, and extracellular signal-regulated kinase 1/2. Circ. Res. 2006, 99, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Lachin, J.M.; McGee, P.; Palmer, J.P. Impact of C-Peptide Preservation on Metabolic and Clinical Outcomes in the Diabetes Control and Complications Trial. Diabetes 2014, 63, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.; Johansson, L.; Wahren, J.; von Bibra, H. C-peptide exerts beneficial effects on myocardial blood flow andfunction in patients with type 1 diabetes. Diabetes 2002, 51, 3077–3082. [Google Scholar] [CrossRef]
- Ido, Y.; Vindigni, A.; Chang, K.; Stramm, L.; Chance, R.; Heath, W.F.; DiMarchi, R.D.; Di Cera, E.; Williamson, J.R. Prevention of vascular and neural dysfunction in diabetic rats by C-peptide. Science 1997, 277, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yorek, M.; Pierson, C.R.; Murakawa, Y.; Breidenbach, A.; Sima, A.A.F. Human C-peptide dose dependently prevents early neuropathy in the BB/Wor-rat. Int. J. Exp. Diabetes Res. 2001, 2, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Faber, O.K.; Hagen, C.; Binder, C. Kinetics of human connecting peptide in normal and diabetic subjects. J. Clin. Investig. 1978, 62, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Foyt, H.; Daniels, M.; Milad, M.; Wahre, J. Pharmacokinetics, safety, and tolerability of a long-acting C-peptide (CBX129801) in patients with type 1 diabetes. Diabetologia 2012, 55, S455. [Google Scholar]
- Jolivalt, C.G.; Rodriguez, M.; Wahren, J.; Calcutt, N.A. Efficacy of a long-acting C-peptide analogue against peripheral neuropathy in streptozotocin-diabetic mice. Diabetes Obes. Metab. 2016, 3, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Wahren, J.; Foyt, H.; Daniels, M.; Arezzo, J.C. Long-acting C-peptide and neuropathy in type 1 diabetes: A 12-month clinical trial. Diabetes Care 2016, 39, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yang, C.; Liu, Q.; Li, J.; Liang, R.; Shen, C.; Zhang, Y.; Wang, K.; Liu, L.; Shezad, K.; et al. Encapsulation of Hydrophilic and Hydrophobic Peptides into Hollow Mesoporous Silica Nanoparticles for Enhancement of Antitumor Immune Response. Small 2017, 13, 1701741. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Na, R.; Wang, X.; Liu, H.; Zhao, L.; Sun, X.; Ma, G.; Cui, F. Fabrication of antimicrobial peptide-loaded PLGA/Chitosan composite microspheres for long-Acting bacterial resistance. Molecules 2017, 22, 1637. [Google Scholar]
- Tang, R.; Wang, X.; Zhang, H.; Liang, X.; Feng, X.; Zhu, X.; Lu, X.; Wu, F.; Liu, Z. Promoting early neovascularization of SIS-repaired abdominal wall by controlled release of bioactive VEGF. RSC Adv. 2018, 8, 4548–4560. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, G.R.; Wakte, P.; Shelke, S. Formulation, physicochemical characterization and in vitro evaluation of human insulin-loaded microspheres as potential oral carrier. Prog. Biomater. 2017, 6, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Maciel, V.B.V.; Yoshida, C.M.P.; Pereira, S.M.S.S.; Goycoolea, F.M.; Franco, T.T. Electrostatic self-assembled chitosan-pectin nano- and microparticles for insulin delivery. Molecules 2017, 22, 1707. [Google Scholar] [CrossRef]
- Bloch, K.; Vanichkin, A.; Gil-Ad, I.; Vardi, P.; Weizman, A. Insulin delivery to the brain using intracranial implantation of alginate-encapsulated pancreatic islets. J. Tissue Eng. Regen. Med. 2017, 11, 3263–3272. [Google Scholar] [CrossRef]
- Alibolandi, M.; Alabdollah, F.; Sadeghi, F.; Mohammadi, M.; Abnous, K.; Ramezani, M.; Hadizadeh, F. Dextran-b-poly (lactide-co-glycolide) polymersome for oral delivery of insulin: In vitro and in vivo evaluation. J. Control. Release 2016, 227, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, S.S.; Malathi, M.S.; Sesh, P.S.L.; Selvasubramanian, S.S.; Balasubramanian, B.S.; Pandiyan, P.V. Hydrophilic poly (ethylene glycol) capped poly (lactic-co-glycolic) acid nanoparticles for subcutaneous delivery of insulin in diabetic rats. Int. J. Biol. Macromol. 2017, 95, 1190–1198. [Google Scholar]
- Gupta, R.; Mohanty, S. Controlled release of insulin from folic acid-insulin complex nanoparticles. Colloids Surf. B Biointerfaces 2017, 154, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Habibi, N.; Kamaly, N.; Memic, A.; Shafiee, H. Self-assembled peptide-based nanostructures: Smart nanomaterials toward targeted drug delivery. Nano Today 2016, 11, 41–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordquist, L.; Palm, F.; Andresen, B.T. Renal and vascular benefits of C-peptide: Molecular mechanisms of C-peptide action. Biologics 2008, 2, 441–452. [Google Scholar] [CrossRef]
- De La Tour, D.; Raccah, D.; Jannot, M.; Coste, T.; Rougerie, C.; Vague, P. Erythrocyte Na/K ATPase activity and diabetes: Relationship with C-peptide level. Diabetologia 1998, 41, 1080–1084. [Google Scholar] [CrossRef]
- Djemli-Shipkolye, A.; Gallice, P.; Coste, T.; Jannot, M.F.; Tsimaratos, M.; Raccah, D.; Vague, P. The effects ex vivo and in vitro of insulin and C-peptide on Na/K adenosine triphosphatase activity in red blood cell membranes of type 1 diabetic patients. Metabolism 2000, 49, 868–872. [Google Scholar] [CrossRef]
- Wilder, R.; Mobashery, S. The use of triphosgene in preparation of N-carboxy-alpha-amino acid anhydrides. J. Org. Chem. 1992, 57, 2755–2756. [Google Scholar] [CrossRef]
- Vlakh, E.; Novikov, A.; Vlasov, G.; Tennikova, T. Solid phase peptide synthesis on epoxy-bearing methacrylate monoliths. J. Pept. Sci. 2004, 10, 719–730. [Google Scholar] [CrossRef]
- Amoabediny, G.; Haghiralsadat, F.; Naderinezhad, S.; Helder, M.N.; Akhoundi Kharanaghi, E.; Mohammadnejad Arough, J.; Zandieh-Doulabi, B. Overview of preparation methods of polymeric and lipid-based (niosome, solid lipid, liposome) nanoparticles: A comprehensive review. Int. J. Polym. Mater. Polym. Biomater. 2018, 67, 383–400. [Google Scholar] [CrossRef]
- Sasaki, Y.; Kohri, M.; Kojima, T.; Taniguchi, T.; Kishikawa, K. Preparation of polymer nanoparticles via phase inversion temperature Method using amphiphilic block polymer synthesized by atom transfer radical polymerization. Trans. Mater. Soc. Jpn. 2014, 39, 125–128. [Google Scholar] [CrossRef]
- Zhu, Y.; Yi, C.; Hu, Q.; Wei, W.; Liu, X. Effect of chain microstructure on self-assembly and emulsification of amphiphilic poly(acrylic acid)-polystyrene copolymers. Phys. Chem. Chem. Phys. 2016, 18, 26236–26244. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Li, W.; Duan, X.; Zhu, L.; Fan, L.; Qiao, Y.; Wu, H. Preparation of two types of polymeric micelles based on poly(β-l-malic acid) for antitumor drug delivery. PLoS ONE 2016, 11, e0162607. [Google Scholar] [CrossRef] [PubMed]
- Prokop, A.; Iwasaki, Y.; Harada, A. (Eds.) Intracellular Delivery II: Fundamentals and Applications; Springer: Heidelberg, Germany, 2014; p. 479. [Google Scholar]
- Marsden, H.R.; Gabrielli, L.; Kros, A. Rapid preparation of polymersomes by a water addition/solvent evaporation method. Polym. Chem. 2010, 1, 1512–1518. [Google Scholar] [CrossRef]
- Tarasenko, I.; Zashikhina, N.; Guryanov, I.; Volokitina, M.; Biondi, B.; Fiorucci, S.; Formaggio, F.; Tennikova, T.; Korzhikova-Vlakh, E. Amphiphilic polypeptides with prolonged enzymatic stability for the preparation of self-assembled nanobiomaterials. RSC Adv. 2018, 8, 34603–34613. [Google Scholar] [CrossRef]
- Zikou, S.; Koukkou, A.-I.; Mastora, P.; Sakarellos-Daitsiotis, M.; Sakarellos, C.; Drainas, C.; Panou-Pomonis, E. Design and synthesis of cationic Aib-containing antimicrobial peptides: Conformational and biological studies. J. Pept. Sci. 2007, 13, 481–486. [Google Scholar] [CrossRef]
- Guo, Q.; Zhang, T.; An, J.; Wu, Z.; Zhao, Y.; Dai, X.; Zhang, X.; Li, C. Block versus Random Amphiphilic Glycopolymer Nanopaticles as Glucose-Responsive Vehicles. Biomacromolecules 2015, 16, 3345–3356. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Ekberg, K.; Shafqat, J.; Henriksson, M.; Chibalin, A.; Wahren, J.; Jörnvall, H. Molecular effects of proinsulin C-peptide. Biochem. Biophys. Res. Commun. 2002, 295, 1035–1040. [Google Scholar] [CrossRef]
- Forst, T.; Kunt, T. Effects of C-peptide on microvascular blood flow and blood hemorheology. Exp. Diabesity Res. 2004, 5, 51–64. [Google Scholar] [CrossRef]
- Jørgensen, P. Sodium and potassium ion pump in kidney tubules. Physiol. Rev. 1980, 60, 864–917. [Google Scholar] [CrossRef] [PubMed]
- Ohtomo, Y.; Bergman, T.; Johansson, B.L.; Jörnvall, H.; Wahren, J. Differential effects of proinsulin C-peptide fragments on Na+, K+- ATPase activity of renal tubule segments. Diabetologia 1998, 41, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Forst, T.; De la Tour, D.D.; Kunt, T.; Pfutzner, A.; Goitom, K.; Pohlmann, T.; Schneider, S.; Johansson, B.L.; Wahren, J.; Lobig, M.; et al. Effects of proinsulin C-peptide on nitric oxide, microvascular blood flow and erythrocyte Na+,K+-atpase activity in diabetes mellitus type I. Clin. Sci. 2000, 98, 283–290. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Initial Ratio of NKAs: [Glu(OBzl)/Lys(Z)]/[d-Phe] | Polymer Characteristics (SEC) | Polymer Yield, % | ||
|---|---|---|---|---|---|
| Mn | Mw | Ð | |||
| P(Glu(OBzl)n-co-dPhem) | |||||
| E(Bzl)F1 | 1/1 | 5600 | 6400 | 1.15 | 49 |
| E(Bzl)F2 | 4/1 | 6700 | 8100 | 1.20 | 70 |
| E(Bzl)F3 | 8/1 | 7100 | 9200 | 1.29 | 72 |
| P(Lys(Z)n-co-dPhem) | |||||
| K(Z)F1 | 1/1 | 14,000 | 15,800 | 1.07 | 68 |
| K(Z)F2 | 4/1 | 21,500 | 24,300 | 1.13 | 55 |
| K(Z)F3 | 8/1 | 24,300 | 28,000 | 1.15 | 71 |
| Sample | Determined Polymer Composition | |||||
|---|---|---|---|---|---|---|
| HPLC | 1H NMR | |||||
| n | m | [Glu/Lys]/[Phe] Ratio | n | m | [Glu]/[Phe] Ratio | |
| P(Glun-co-dPhem) | ||||||
| EF1 | 17 | 14 | 1.2 | 16 | 15 | 1.1 |
| EF2 | 33 | 11 | 3.0 | 37 | 10 | 3.7 |
| EF3 | 38 | 7 | 5.4 | 45 | 8 | 5.6 |
| P(Lysn-co-dPhem) | ||||||
| KF1 | 34 | 34 | 1.0 | - | - | - |
| KF2 | 72 | 17 | 4.3 | - | - | - |
| KF3 | 87 | 9 | 9.5 | - | - | - |
| Sample | Amount of Bound Peptide, μg/mg of Nanospheres | Amount of Bound Peptide, nmol/mg of Nanospheres | Immobilization Efficiency, % |
|---|---|---|---|
| C-peptide | |||
| EF1 | 20 ± 4 | 5.5 | 10 ± 1 |
| EF2 | 48 ± 5 | 13.3 | 24 ± 2 |
| EF3 | 25 ± 3 | 6.9 | 13 ± 2 |
| C5 | |||
| EF2 | 16 ± 2 | 30.0 | 16 ± 2 |
| Sample | DHo *, nm | EE **, % | LC, µg/mg of Particles | DH encaps *, nm | PDI encaps * |
|---|---|---|---|---|---|
| C-peptide | |||||
| KF1 | 71 ± 3 | 89.5 ± 0.6 | 89.5 ± 0.5 | 79 ± 8 | 0.24 |
| KF2 | 96 ± 3 | 94.6 ± 1.0 | 94.6 ± 0.9 | 130 ± 20 | 0.16 |
| KF3 | 150 ± 10 | 95.0 ± 1.1 | 95.0 ± 1.0 | 190 ± 20 | 0.14 |
| C5 | |||||
| KF3 | 150 ± 10 | 96.2 ± 0.7 | 96.2 ± 0.7 | 178 ± 15 | 0.13 |
| Sample | Amount of C-Peptide Encapsulated, µg/mg of Nanospheres | Amount of C-Peptide Retained after 14 Days Release, µg/mg of Nanospheres |
|---|---|---|
| KF1 | 581 ± 3 | 157 ± 4 |
| KF3-HLD | 604 ± 4 | 259 ± 5 |
| KF3-MLD | 285 ± 5 | 225 ± 7 |
| KF3-LLD | 147 ± 4 | 128 ± 3 |
| KF3-HLD + heparin | 483 ± 8 | 335 ± 10 |
| # | Experimental Condition | ∆H, μJ Normalized to Control |
|---|---|---|
| 1 | C-peptide + ouabain | 0 * |
| 2 | C-peptide | −102 ± 7 * |
| 3 | C5 | −136 ± 9 * |
| 4 | C-peptide encapsulated in KF3 nanospheres (HLD) | −265 ± 19 ** |
| 5 | C-peptide encapsulated in KF3 nanospheres (LLD) | −65 ± 5 ** |
| 6 | C-peptide immobilized on the surface of EF2 nanospheres | −213 ± 16 *** |
| 7 | C5 encapsulated in KF3 nanospheres (LLD) | −54 ± 6 ** |
| 8 | C5 immobilized on the surface of EF2 nanospheres | −15 ± 4 *** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zashikhina, N.; Sharoyko, V.; Antipchik, M.; Tarasenko, I.; Anufrikov, Y.; Lavrentieva, A.; Tennikova, T.; Korzhikova-Vlakh, E. Novel Formulations of C-Peptide with Long-Acting Therapeutic Potential for Treatment of Diabetic Complications. Pharmaceutics 2019, 11, 27. https://doi.org/10.3390/pharmaceutics11010027
Zashikhina N, Sharoyko V, Antipchik M, Tarasenko I, Anufrikov Y, Lavrentieva A, Tennikova T, Korzhikova-Vlakh E. Novel Formulations of C-Peptide with Long-Acting Therapeutic Potential for Treatment of Diabetic Complications. Pharmaceutics. 2019; 11(1):27. https://doi.org/10.3390/pharmaceutics11010027
Chicago/Turabian StyleZashikhina, Natalia, Vladimir Sharoyko, Mariia Antipchik, Irina Tarasenko, Yurii Anufrikov, Antonina Lavrentieva, Tatiana Tennikova, and Evgenia Korzhikova-Vlakh. 2019. "Novel Formulations of C-Peptide with Long-Acting Therapeutic Potential for Treatment of Diabetic Complications" Pharmaceutics 11, no. 1: 27. https://doi.org/10.3390/pharmaceutics11010027
APA StyleZashikhina, N., Sharoyko, V., Antipchik, M., Tarasenko, I., Anufrikov, Y., Lavrentieva, A., Tennikova, T., & Korzhikova-Vlakh, E. (2019). Novel Formulations of C-Peptide with Long-Acting Therapeutic Potential for Treatment of Diabetic Complications. Pharmaceutics, 11(1), 27. https://doi.org/10.3390/pharmaceutics11010027