Drug Bioavailability Enhancing Agents of Natural Origin (Bioenhancers) that Modulate Drug Membrane Permeation and Pre-Systemic Metabolism

 ,
, 

Abstract
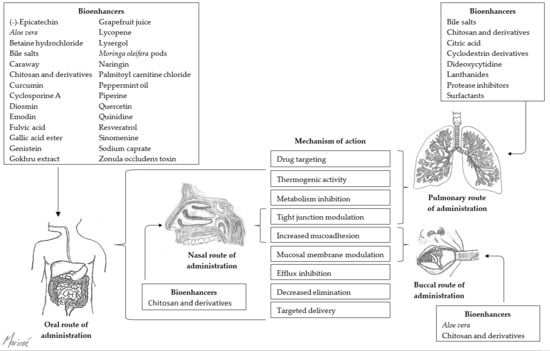
:
1. Introduction
2. Buccal Route of Administration
2.1. Aloe Vera
2.2. Bile Salts
2.3. Chitosan and Derivatives
2.4. Fatty Acids
2.5. Menthol
3. Nasal Route of Administration
3.1. Bile Salts
3.2. Chitosan and Derivatives
3.3. Starch Microspheres
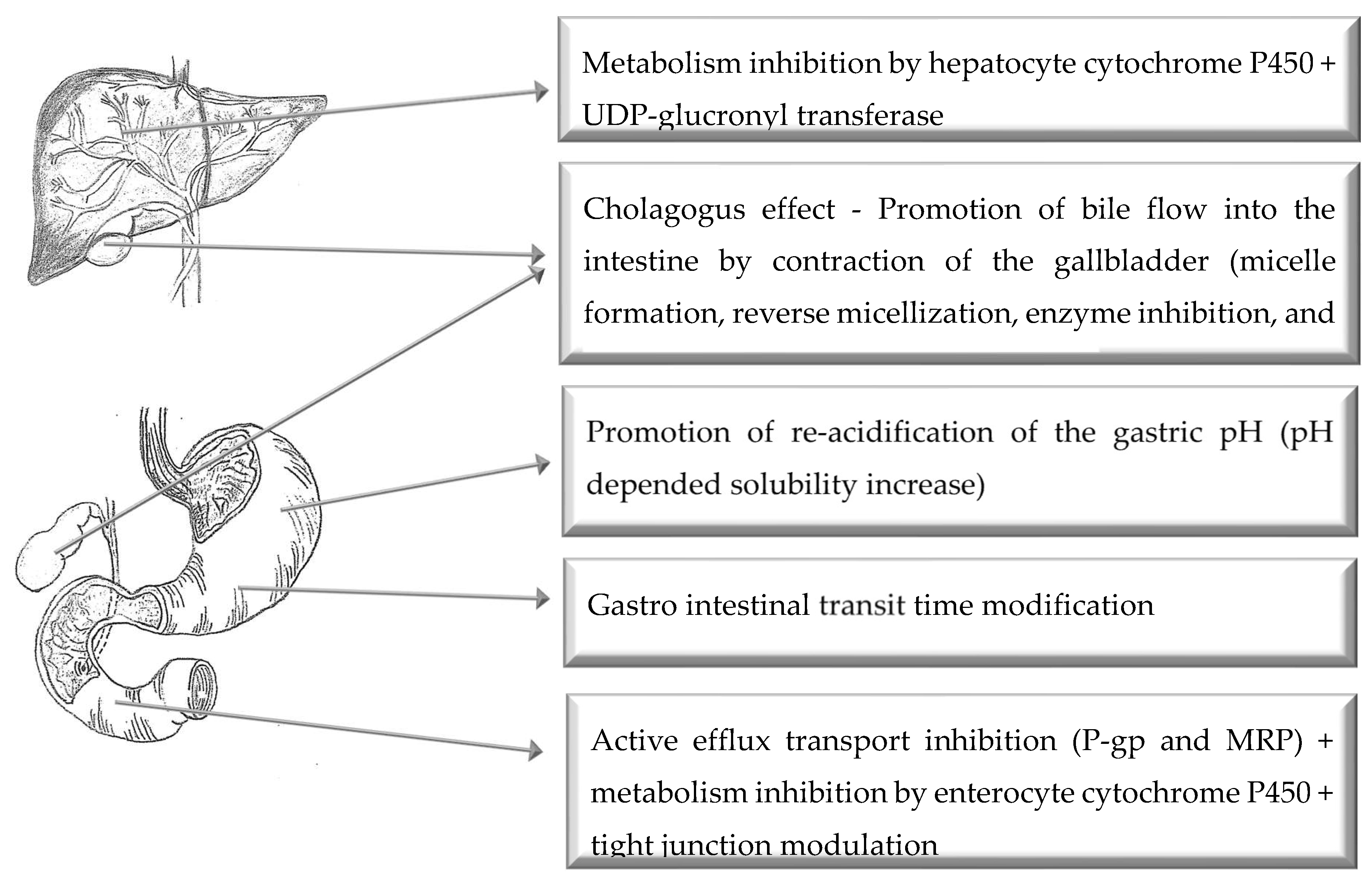
4. Oral Route of Administration
4.1. Aloe Vera
4.2. Bile Salts
4.3. Black Cumin
4.4. Capsaicin
4.5. Caraway
4.6. Cylcosporine A
4.7. Chitosan and Derivatives
4.8. Curcumin
4.9. Diosmin
4.10. Emodin
4.11. Gallic Acid Ester
4.12. Genistein
4.13. Gokhru Extract
4.14. Grapefruit Juice
4.15. Lycopene
4.16. Lysergol
4.17. Naringin and Bergamottin
4.18. Palmitoyl Carnitine Chloride
4.19. Piperine
4.20. Quercetin
4.21. Quinidine
4.22. Resveratrol
4.23. Sinomenine
4.24. Sodium Caprate (Fatty Acid)
4.25. Zonula Occludens Toxin (Zot)
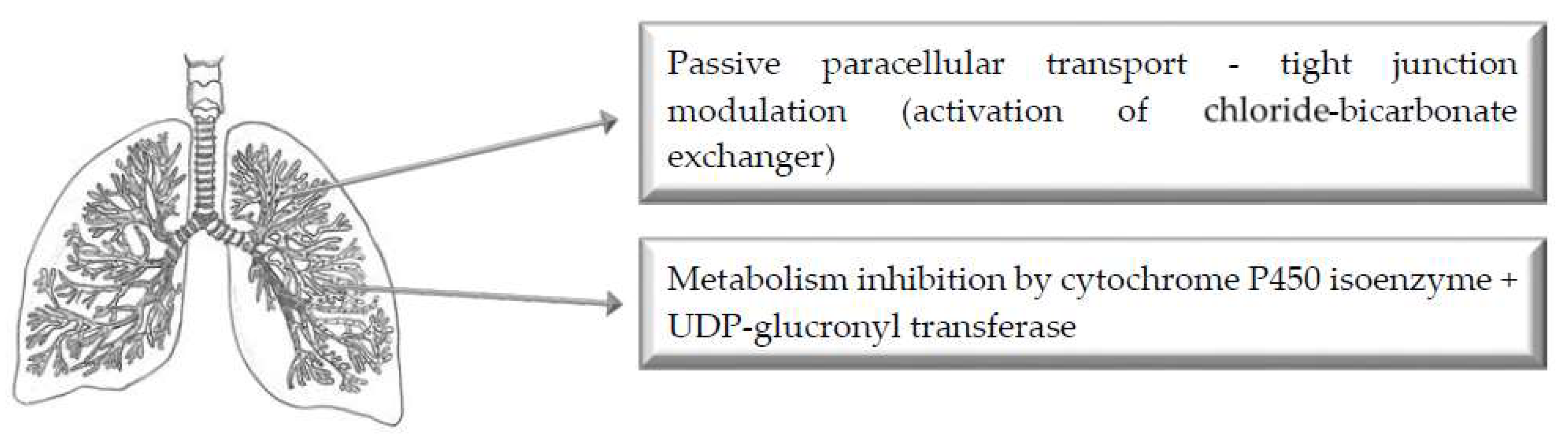
5. Pulmonary Route of Administration
5.1. Bile Salts
5.2. Chitosan and Derivatives
5.3. Citric Acid (Chelating Agent)
5.4. Cyclodextrins (CDs)
5.5. Lanthanides
5.6. Protease Inhibitors
5.7. Surfactants
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Navia, M.A.; Chaturvedi, P.R. Design principles for orally bioavailable drugs. Drug Discov. Today 1996, 1, 179–189. [Google Scholar] [CrossRef]
- Chillistone, S.; Hardman, J. Factors affecting drug absorption and distribution. Anaesth. Intensive Care Med. 2008, 9, 167–171. [Google Scholar] [CrossRef]
- Klein, I.; Sarkadi, B.; Váradi, A. An inventory of the human abc proteins. Biochim. Biophys. Acta 1999, 1461, 237–262. [Google Scholar] [CrossRef]
- Higgins, C.F.; Gottesman, M.M. Is the multidrug transporter a flippase? Trends Biochem. Sci. 1992, 17, 18–21. [Google Scholar] [CrossRef]
- Sikic, B.I. Modulation of multidrug resistance: A paradigm for translational clinical research. Oncology 1999, 13, 183–187. [Google Scholar] [PubMed]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Maliepaard, M.; Scheffer, G.L.; Faneyte, I.F.; van Gastelen, M.A.; Pijnenborg, A.C.; Schinkel, A.H.; van de Vijver, M.J.; Scheper, R.J.; Schellens, J.H. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001, 61, 3458–3464. [Google Scholar]
- Lazzari, P.; Serra, V.; Marcello, S.; Pira, M.; Mastinu, A. Metabolic side effects induced by olanzapine treatment are neutralized by cb1 receptor antagonist compounds co-administration in female rats. Eur. Neuropsychopharmacol. 2017, 27, 667–678. [Google Scholar] [CrossRef]
- Dresser, G.K.; Spence, J.D.; Bailey, D.G. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome p450 3a4 inhibition. Clin. Pharmacokinet. 2000, 38, 41–57. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the atp binding cassette (abc) family: An overview. Adv. Drug Deliv. Rev. 2012, 64, 138–153. [Google Scholar] [CrossRef]
- Khajuria, A.; Thusu, N.; Zutshi, U. Piperine modulates permeability characteristics of intestine by inducing alterations in membrane dynamics: Influence on brush border membrane fluidity, ultrastructure and enzyme kinetics. Phytomedicine 2002, 9, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Sarangi, M.; Chandra-Joshi, B.; Ritchie, B. Natural bioenhancers in drug delivery: An overview. Puerto Rico Health Sci. J. 2018, 37, 12–18. [Google Scholar]
- Bitter, C.; Suter-Zimmermann, K.; Surber, C. Nasal drug delivery in humans. Curr. Probl. Dermatol. 2011, 40, 20–35. [Google Scholar] [PubMed]
- Liu, Z.; Wang, S.; Hu, M. Oral absorption basics: Pathways, physico-chemical and biological factors affecting absorption. In Developing Solid Oral Dosage Forms; Elsevier: Amsterdam, The Netherlands, 2009; pp. 263–288. [Google Scholar]
- Crowley, P.; Martini, L. Optimising drug delivery: The challenges and opportunities. Ondrugdelivery 2015, 2015, 4–11. [Google Scholar]
- Mastinu, A.; Premoli, M.; Maccarinelli, G.; Grilli, M.; Memo, M.; Bonini, S.A. Melanocortin 4 receptor stimulation improves social deficits in mice through oxytocin pathway. Neuropharmacology 2018, 133, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.A. Drug metabolism in the nasal mucosa. Pharm. Res. 1992, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.H.; Quay, S.C. Advances in nasal drug delivery through tight junction technology. Expert Opin. Drug Deliv. 2005, 2, 281–298. [Google Scholar] [CrossRef]
- Bonini, S.A.; Premoli, M.; Tambaro, S.; Kumar, A.; Maccarinelli, G.; Memo, M.; Mastinu, A. Cannabis sativa: A comprehensive ethnopharmacological review of a medicinal plant with a long history. J. Ethnopharmacol. 2018, 227, 300–315. [Google Scholar] [CrossRef]
- Ojewole, E.; Mackraj, I.; Akhundov, K.; Hamman, J.; Viljoen, A.; Olivier, E.; Wesley-Smith, J.; Govender, T. Investigating the effect of aloe vera gel on the buccal permeability of didanosine. Planta Med. 2012, 78, 354–361. [Google Scholar] [CrossRef]
- Portero, A.; Remuñán-López, C.; Nielsen, H.M. The potential of chitosan in enhancing peptide and protein absorption across the tr146 cell culture model—An in vitro model of the buccal epithelium. Pharm. Res. 2002, 19, 169–174. [Google Scholar] [CrossRef]
- Şenel, S.; Kremer, M.; Kaş, S.; Wertz, P.; Hıncal, A.; Squier, C. Enhancing effect of chitosan on peptide drug delivery across buccal mucosa. Biomaterials 2000, 21, 2067–2071. [Google Scholar] [CrossRef]
- Langoth, N.; Kahlbacher, H.; Schöffmann, G.; Schmerold, I.; Schuh, M.; Franz, S.; Kurka, P.; Bernkop-Schnürch, A. Thiolated chitosans: Design and in vivo evaluation of a mucoadhesive buccal peptide drug delivery system. Pharm. Res. 2006, 23, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Langoth, N.; Bernkop-Schnürch, A.; Kurka, P. In vitro evaluation of various buccal permeation enhancing systems for pacap (pituitary adenylate cyclase-activating polypeptide). Pharm. Res. 2005, 22, 2045. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, K.; Obata, Y.; Takayama, K.; Loftsson, T.; Nagai, T. Effect of cod-liver oil extract on the buccal permeation of ergotamine tartrate. Drug Dev. Ind. Pharm. 1998, 24, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, A.H.; Khan, M.; Lim, G.; Khosravan, R. Transbuccal permeation of a nucleoside analog, dideoxycytidine: Effects of menthol as a permeation enhancer. Int. J. Pharm. 1999, 192, 139–146. [Google Scholar] [CrossRef]
- Morishita, M.; Barichello, J.M.; Takayama, K.; Chiba, Y.; Tokiwa, S.; Nagai, T. Pluronic® f-127 gels incorporating highly purified unsaturated fatty acids for buccal delivery of insulin. Int. J. Pharm. 2001, 212, 289–293. [Google Scholar] [CrossRef]
- Xiang, J.; Fang, X.; Li, X. Transbuccal delivery of 2′, 3′-dideoxycytidine: In vitro permeation study and histological investigation. Int. J. Pharm. 2002, 231, 57–66. [Google Scholar] [CrossRef]
- Sandri, G.; Rossi, S.; Bonferoni, M.C.; Ferrari, F.; Zambito, Y.; Di Colo, G.; Caramella, C. Buccal penetration enhancement properties of n-trimethyl chitosan: Influence of quaternization degree on absorption of a high molecular weight molecule. Int. J. Pharm. 2005, 297, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Hinchcliffe, M.; Jabbal-Gill, I.; Smith, A. Effect of chitosan on the intranasal absorption of salmon calcitonin in sheep. J. Pharm. Pharmacol. 2005, 57, 681–687. [Google Scholar] [CrossRef]
- Illum, L.; Watts, P.; Fisher, A.; Hinchcliffe, M.; Norbury, H.; Jabbal-Gill, I.; Nankervis, R.; Davis, S. Intranasal delivery of morphine. J. Pharmacol. Exp. Ther. 2002, 301, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Krauland, A.H.; Guggi, D.; Bernkop-Schnürch, A. Thiolated chitosan microparticles: A vehicle for nasal peptide drug delivery. Int. J. Pharm. 2006, 307, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Hamman, J.; Stander, M.; Kotze, A. Effect of the degree of quaternisation of N-trimethyl chitosan chloride on absorption enhancement: In vivo evaluation in rat nasal epithelia. Int. J. Pharm. 2002, 232, 235–242. [Google Scholar] [CrossRef]
- Mizuma, T.; Awazu, S. Dietary polyphenols (−)-epicatechin and chrysin inhibit intestinal glucuronidation metabolism to increase drug absorption. J. Pharm. Sci. 2004, 93, 2407–2410. [Google Scholar] [CrossRef] [PubMed]
- Beneke, C.; Viljoen, A.; Hamman, J.H. In vitro drug absorption enhancement effects of aloe vera and aloe ferox. Sci Pharm. 2012, 80, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lu, Z.; Viljoen, A.; Hamman, J. Intestinal drug transport enhancement by aloe vera. Planta Med. 2009, 75, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Vinson, J.A.; Al Kharrat, H.; Andreoli, L. Effect of aloe vera preparations on the human bioavailability of vitamins c and e. Phytomedicine 2005, 12, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Wallis, L.; Malan, M.; Gouws, C.; Steyn, D.; Ellis, S.; Abay, E.; Wiesner, L.; P Otto, D.; Hamman, J. Evaluation of isolated fractions of aloe vera gel materials on indinavir pharmacokinetics: In vitro and in vivo studies. Curr. Drug Deliv. 2016, 13, 471–480. [Google Scholar] [CrossRef]
- Yago, M.R.; Frymoyer, A.; Benet, L.Z.; Smelick, G.S.; Frassetto, L.A.; Ding, X.; Dean, B.; Salphati, L.; Budha, N.; Jin, J.Y. The use of betaine hcl to enhance dasatinib absorption in healthy volunteers with rabeprazole-induced hypochlorhydria. AAPS J. 2014, 16, 1358–1365. [Google Scholar] [CrossRef]
- Choudhary, N.; Khajuria, V.; Gillani, Z.H.; Tandon, V.R.; Arora, E. Effect of carum carvi, a herbal bioenhancer on pharmacokinetics of antitubercular drugs: A study in healthy human volunteers. Perspect. Clin. Res. 2014, 5, 80. [Google Scholar]
- Rosenthal, R.; Günzel, D.; Finger, C.; Krug, S.M.; Richter, J.F.; Schulzke, J.-D.; Fromm, M.; Amasheh, S. The effect of chitosan on transcellular and paracellular mechanisms in the intestinal epithelial barrier. Biomaterials 2012, 33, 2791–2800. [Google Scholar] [CrossRef]
- Schipper, N.G.; Olsson, S.; Hoogstraate, J.A.; Vårum, K.M.; Artursson, P. Chitosans as absorption enhancers for poorly absorbable drugs 2: Mechanism of absorption enhancement. Pharm. Res. 1997, 14, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Bernkop-Schnürch, A.; Guggi, D.; Pinter, Y. Thiolated chitosans: Development and in vitro evaluation of a mucoadhesive, permeation enhancing oral drug delivery system. J. Control. Release 2004, 94, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Krauland, A.H.; Guggi, D.; Bernkop-Schnürch, A. Oral insulin delivery: The potential of thiolated chitosan-insulin tablets on non-diabetic rats. J. Control. Release 2004, 95, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Basu, N.K.; Kole, L.; Kubota, S.; Owens, I.S. Human udp-glucuronosyltransferases show atypical metabolism of mycophenolic acid and inhibition by curcumin. Drug Metab. Dispos. 2004, 32, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Pavithra, B.; Prakash, N.; Jayakumar, K. Modification of pharmacokinetics of norfloxacin following oral administration of curcumin in rabbits. J. Vet. Sci. 2009, 10, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lim, L.-Y. Effects of spice constituents on p-gp-mediated transport and cyp3a4-mediated metabolism in vitro. Drug Metab. Dispos. 2008, 36, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tan, T.M.C.; Lim, L.-Y. Impact of curcumin-induced changes in p-glycoprotein and cyp3a expression on the pharmacokinetics of peroral celiprolol and midazolam in rats. Drug Metab. Dispos. 2007, 35, 110–115. [Google Scholar] [CrossRef]
- Lee, J.H.; Shin, Y.-J.; Oh, J.-H.; Lee, Y.-J. Pharmacokinetic interactions of clopidogrel with quercetin, telmisartan, and cyclosporine a in rats and dogs. Arch. Pharm. Res. 2012, 35, 1831–1837. [Google Scholar] [CrossRef]
- Yoo, H.H.; Lee, M.; Chung, H.J.; Lee, S.K.; Kim, D.-H. Effects of diosmin, a flavonoid glycoside in citrus fruits, on p-glycoprotein-mediated drug efflux in human intestinal caco-2 cells. J. Agric. Food Chem. 2007, 55, 7620–7625. [Google Scholar] [CrossRef]
- Li, X.; Hu, J.; Wang, B.; Sheng, L.; Liu, Z.; Yang, S.; Li, Y. Inhibitory effects of herbal constituents on p-glycoprotein in vitro and in vivo: Herb–drug interactions mediated via p-gp. Toxicol. Appl. Pharmacol. 2014, 275, 163–175. [Google Scholar] [CrossRef]
- Ghosal, S. Delivery System for Pharmaceutical, Nutritional and Cosmetic Ingredients. U.S. Patent 6,558,712, 6 May 2003. [Google Scholar]
- Wacher, V.J.; Benet, L.Z. Use of Gallic Acid Esters to Increase Bioavailability of Orally Administered Pharmaceutical Compounds. U.S. Patent 5,962,522, 5 October 1999. [Google Scholar]
- Lambert, J.D.; Kwon, S.-J.; Ju, J.; Bose, M.; Lee, M.-J.; Hong, J.; Hao, X.; Yang, C.S. Effect of genistein on the bioavailability and intestinal cancer chemopreventive activity of (−)-epigallocatechin-3-gallate. Carcinogenesis 2008, 29, 2019–2024. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Choi, J.-S. Effect of genistein on the pharmacokinetics of paclitaxel administered orally or intravenously in rats. Int. J. Pharm. 2007, 337, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Ayyanna, C.; Mohan Rao, G.; Sasikala, M.; Somasekhar, P.; Arun Kumar, N.; Pradeep Kumar, M. Absorption enhancement studies of metformin hydrochloride by using tribulus terrestris plant extract. Int. J. Pharm. Technol. 2012, 4, 4119–4125. [Google Scholar]
- Kumar, A.; Bansal, M. Formulation and evaluation of antidiabetic tablets: Effect of absorption enhancser. World J. Pharm. Res. 2014, 3, 1426–1445. [Google Scholar]
- Dresser, G.; Bailey, D. The effects of fruit juices on drug disposition: A new model for drug interactions. Eur. J. Clin. Investig. 2003, 33, 10–16. [Google Scholar] [CrossRef]
- Rekha, M.; Sharma, C.P. Synthesis and evaluation of lauryl succinyl chitosan particles towards oral insulin delivery and absorption. J. Control. Release 2009, 135, 144–151. [Google Scholar] [CrossRef]
- Petyaev, I.M. Improvement of hepatic bioavailability as a new step for the future of statin. Arch. Med Sci. 2015, 11, 406. [Google Scholar] [CrossRef]
- Patil, S.; Dash, R.P.; Anandjiwala, S.; Nivsarkar, M. Simultaneous quantification of berberine and lysergol by hplc-uv: Evidence that lysergol enhances the oral bioavailability of berberine in rats. Biomed. Chromatogr. 2012, 26, 1170–1175. [Google Scholar] [CrossRef]
- Shukla, M.; Malik, M.; Jaiswal, S.; Sharma, A.; Tanpula, D.; Goyani, R.; Lal, J. A mechanistic investigation of the bioavailability enhancing potential of lysergol, a novel bioenhancer, using curcumin. RSC Adv. 2016, 6, 58933–58942. [Google Scholar] [CrossRef]
- Pal, A.; Bawankule, D.U.; Darokar, M.P.; Gupta, S.C.; Arya, J.S.; Shanker, K.; Gupta, M.M.; Yadav, N.P.; Singh Khanuja, S.P. Influence of moringa oleifera on pharmacokinetic disposition of rifampicin using hplc-pda method: A pre-clinical study. Biomed. Chromatogr. 2011, 25, 641–645. [Google Scholar] [CrossRef]
- Choi, J.-S.; Han, H.-K. Enhanced oral exposure of diltiazem by the concomitant use of naringin in rats. Int. J. Pharm. 2005, 305, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Kang, K.W. Enhanced tamoxifen bioavailability after oral administration of tamoxifen in rats pretreated with naringin. Arch. Pharm. Res. 2008, 31, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Shin, S.-C. Enhanced paclitaxel bioavailability after oral coadministration of paclitaxel prodrug with naringin to rats. Int. J. Pharm. 2005, 292, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Lassoued, M.A.; Sfar, S.; Bouraoui, A.; Khemiss, F. Absorption enhancement studies of clopidogrel hydrogen sulphate in rat everted gut sacs. J. Pharm. Pharmacol. 2012, 64, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Yeum, C.-H.; Choi, J.-S. Effect of naringin pretreatment on bioavailability of verapamil in rabbits. Arch. Pharm. Res. 2006, 29, 102. [Google Scholar] [CrossRef] [PubMed]
- Raiman, J.; Törmälehto, S.; Yritys, K.; Junginger, H.E.; Mönkkönen, J. Effects of various absorption enhancers on transport of clodronate through caco-2 cells. Int. J. Pharm. 2003, 261, 129–136. [Google Scholar] [CrossRef]
- Wacher, V.J.; Wong, S.; Wong, H.T. Peppermint oil enhances cyclosporine oral bioavailability in rats: Comparison with d-α-tocopheryl poly (ethylene glycol 1000) succinate (tpgs) and ketoconazole. J. Pharm. Sci. 2002, 91, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Badmaev, V.; Majeed, M.; Norkus, E.P. Piperine, an alkaloid derived from black pepper increases serum response of β-carotene during 14-days of oral β-carotene supplementation. Nutr. Res. 1999, 19, 381–388. [Google Scholar] [CrossRef]
- Badmaev, V.; Majeed, M.; Prakash, L. Piperine derived from black pepper increases the plasma levels of coenzyme q10 following oral supplementation. J. Nutr. Biochem. 2000, 11, 109–113. [Google Scholar] [CrossRef]
- Bajad, S.; Bedi, K.; Singla, A.; Johri, R. Piperine inhibits gastric emptying and gastrointestinal transit in rats and mice. Planta Med. 2001, 67, 176–179. [Google Scholar] [CrossRef]
- Bano, G.; Raina, R.; Zutshi, U.; Bedi, K.; Johri, R.; Sharma, S. Effect of piperine on bioavailability and pharmacokinetics of propranolol and theophylline in healthy volunteers. Eur. J. Clin. Pharmacol. 1991, 41, 615–617. [Google Scholar] [CrossRef]
- Gupta, S.; Bansal, P.; Bhardwaj, R.; Velpandian, T. Comparative anti-nociceptive, anti-inflammatory and toxicity profile of nimesulide vs nimesulide and piperine combination. Pharmacol. Res. 2000, 41, 657–662. [Google Scholar] [CrossRef]
- Jin, M.J.; Han, H.K. Effect of piperine, a major component of black pepper, on the intestinal absorption of fexofenadine and its implication on food–drug interaction. J. Food Sci. 2010, 75, H93–H96. [Google Scholar] [CrossRef]
- Johnson, J.J.; Nihal, M.; Siddiqui, I.A.; Scarlett, C.O.; Bailey, H.H.; Mukhtar, H.; Ahmad, N. Enhancing the bioavailability of resveratrol by combining it with piperine. Mol. Nutr. Food Res. 2011, 55, 1169–1176. [Google Scholar] [CrossRef] [Green Version]
- Kasibhatta, R.; Naidu, M. Influence of piperine on the pharmacokinetics of nevirapine under fasting conditions. Drugs R D 2007, 8, 383–391. [Google Scholar] [CrossRef]
- Lambert, J.D.; Hong, J.; Kim, D.H.; Mishin, V.M.; Yang, C.S. Piperine enhances the bioavailability of the tea polyphenol (−)-epigallocatechin-3-gallate in mice. J. Nutr. 2004, 134, 1948–1952. [Google Scholar] [CrossRef]
- Mujumdar, A.; Dhuley, J.; Deshmukh, V.; Raman, P.; Thorat, S.; Naik, S. Effect of piperine on pentobarbitone induced hypnosis in rats. Indian J. Exp. Boil. 1990, 28, 486–487. [Google Scholar]
- Pattanaik, S.; Hota, D.; Prabhakar, S.; Kharbanda, P.; Pandhi, P. Pharmacokinetic interaction of single dose of piperine with steady-state carbamazepine in epilepsy patients. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2009, 23, 1281–1286. [Google Scholar] [CrossRef]
- Sama, V.; Nadipelli, M.; Yenumula, P.; Bommineni, M.; Mullangi, R. Effect of piperine on antihyperglycemic activity and pharmacokinetic profile of nateglinide. Arzneimittelforschung 2012, 62, 384–388. [Google Scholar] [CrossRef]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef]
- Singh, M.; Varshneya, C.; Telang, R.; Srivastava, A. Alteration of pharmacokinetics of oxytetracycline following oral administration of piper longum in hens. J. Vet. Sci. 2005, 6, 197–200. [Google Scholar]
- Babu, P.R.; Babu, K.N.; Peter, P.H.; Rajesh, K.; Babu, P.J. Influence of quercetin on the pharmacokinetics of ranolazine in rats and in vitro models. Drug Dev. Ind. Pharm. 2013, 39, 873–879. [Google Scholar] [CrossRef]
- Bansal, T.; Awasthi, A.; Jaggi, M.; Khar, R.K.; Talegaonkar, S. Pre-clinical evidence for altered absorption and biliary excretion of irinotecan (cpt-11) in combination with quercetin: Possible contribution of p-glycoprotein. Life Sci. 2008, 83, 250–259. [Google Scholar] [CrossRef]
- Challa, V.R.; Ravindra Babu, P.; Challa, S.R.; Johnson, B.; Maheswari, C. Pharmacokinetic interaction study between quercetin and valsartan in rats and in vitro models. Drug Dev. Ind. Pharm. 2013, 39, 865–872. [Google Scholar] [CrossRef]
- Choi, J.S.; Han, H.K. The effect of quercetin on the pharmacokinetics of verapamil and its major metabolite, norverapamil, in rabbits. J. Pharm. Pharmacol. 2004, 56, 1537–1542. [Google Scholar] [CrossRef]
- Choi, J.-S.; Li, X. Enhanced diltiazem bioavailability after oral administration of diltiazem with quercetin to rabbits. Int. J. Pharm. 2005, 297, 1–8. [Google Scholar] [CrossRef]
- Choi, J.-S.; Piao, Y.-J.; Kang, K.W. Effects of quercetin on the bioavailability of doxorubicin in rats: Role of cyp3a4 and p-gp inhibition by quercetin. Arch. Pharm. Res. 2011, 34, 607–613. [Google Scholar] [CrossRef]
- Kim, K.-A.; Park, P.-W.; Park, J.-Y. Short-term effect of quercetin on the pharmacokinetics of fexofenadine, a substrate of p-glycoprotein, in healthy volunteers. Eur. J. Clin. Pharmacol. 2009, 65, 609–614. [Google Scholar] [CrossRef]
- Li, X.; Choi, J.-S. Effects of quercetin on the pharmacokinetics of etoposide after oral or intravenous administration of etoposide in rats. Anticancer. Res. 2009, 29, 1411–1415. [Google Scholar]
- Nijveldt, R.J.; Van Nood, E.; Van Hoorn, D.E.; Boelens, P.G.; Van Norren, K.; Van Leeuwen, P.A. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [CrossRef]
- Scambia, G.; Ranelletti, F.; Panici, P.B.; De Vincenzo, R.; Bonanno, G.; Ferrandina, G.; Piantelli, M.; Bussa, S.; Rumi, C.; Cianfriglia, M. Quercetin potentiates the effect of adriamycin in a multidrug-resistant mcf-7 human breast-cancer cell line: P-glycoprotein as a possible target. Cancer Chemother. Pharmacol. 1994, 34, 459–464. [Google Scholar] [CrossRef]
- Shin, S.-C.; Choi, J.-S.; Li, X. Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int. J. Pharm. 2006, 313, 144–149. [Google Scholar] [CrossRef]
- Umathe, S.N.; Dixit, P.V.; Bansod, K.U.; Wanjari, M.M. Quercetin pretreatment increases the bioavailability of pioglitazone in rats: Involvement of cyp3a inhibition. Biochem. Pharmacol. 2008, 75, 1670–1676. [Google Scholar] [CrossRef]
- Chan, K.; Liu, Z.Q.; Jiang, Z.H.; Zhou, H.; Wong, Y.F.; Xu, H.-X.; Liu, L. The effects of sinomenine on intestinal absorption of paeoniflorin by the everted rat gut sac model. J. Ethnopharmacol. 2006, 103, 425–432. [Google Scholar] [CrossRef]
- Bedada, S.K.; Yellu, N.R.; Neerati, P. Effect of resveratrol treatment on the pharmacokinetics of diclofenac in healthy human volunteers. Phytother. Res. 2016, 30, 397–401. [Google Scholar] [CrossRef]
- Jia, Y.; Liu, Z.; Wang, C.; Meng, Q.; Huo, X.; Liu, Q.; Sun, H.; Sun, P.; Yang, X.; Ma, X. P-gp, mrp2 and oat1/oat3 mediate the drug-drug interaction between resveratrol and methotrexate. Toxicol. Appl. Pharmacol. 2016, 306, 27–35. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Zhou, H.; Liu, L.; Jiang, Z.H.; Wong, Y.F.; Xie, Y.; Cai, X.; Xu, H.X.; Chan, K. Influence of co-administrated sinomenine on pharmacokinetic fate of paeoniflorin in unrestrained conscious rats. J. Ethnopharmacol. 2005, 99, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.-Y.; Li, J.; Zhang, M.; Wang, C.-M.; Fan, Z.; Wang, C.-Y.; Chen, L. Enhancement of sodium caprate on intestine absorption and antidiabetic action of berberine. AAPS Pharm. 2010, 11, 372–382. [Google Scholar] [CrossRef]
- Yu, J.-N.; Zhu, Y.; Wang, L.; Peng, M.; Tong, S.-S.; Cao, X.; Qiu, H.; Xu, X.-M. Enhancement of oral bioavailability of the poorly water-soluble drug silybin by sodium cholate/phospholipid-mixed micelles. Acta Pharmacol. Sin. 2010, 31, 759. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, Y.; Chen, J.; Lai, J.; Sun, J.; Hu, F.; Wu, W. Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt. Int. J. Pharm. 2009, 376, 153–160. [Google Scholar] [CrossRef]
- Wang, H.-J.; Pao, L.-H.; Hsiong, C.-H.; Shih, T.-Y.; Lee, M.-S.; Hu, O.Y.-P. Dietary flavonoids modulate cyp2c to improve drug oral bioavailability and their qualitative/quantitative structure–activity relationship. AAPS J. 2014, 16, 258–268. [Google Scholar] [CrossRef]
- Hamman, J.; Schultz, C.; Kotzé, A. N-trimethyl chitosan chloride: Optimum degree of quaternization for drug absorption enhancement across epithelial cells. Drug Dev. Ind. Pharm. 2003, 29, 161–172. [Google Scholar] [CrossRef]
- Kotzé, A.R.; Lueβen, H.L.; de Leeuw, B.J.; Verhoef, J.C.; Junginger, H.E. N-trimethyl chitosan chloride as a potential absorption enhancer across mucosal surfaces: In vitro evaluation in intestinal epithelial cells (caco-2). Pharm. Res. 1997, 14, 1197–1202. [Google Scholar] [CrossRef]
- Cox, D.S.; Raje, S.; Gao, H.; Salama, N.N.; Eddington, N.D. Enhanced permeability of molecular weight markers and poorly bioavailable compounds across caco-2 cell monolayers using the absorption enhancer, zonula occludens toxin. Pharm. Res. 2002, 19, 1680–1688. [Google Scholar] [CrossRef]
- Machida, M.; Hayashi, M.; Awazu, S. The effects of absorption enhancers on the pulmonary absorption of recombinant human granulocyte colony-stimulating factor (rhg-csf) in rats. Boil. Pharm. Bull. 2000, 23, 84–86. [Google Scholar] [CrossRef]
- Florea, B.I.; Thanou, M.; Junginger, H.E.; Borchard, G. Enhancement of bronchial octreotide absorption by chitosan and n-trimethyl chitosan shows linear in vitro/in vivo correlation. J. Control. Release 2006, 110, 353–361. [Google Scholar] [CrossRef]
- Todo, H.; Okamoto, H.; Iida, K.; Danjo, K. Effect of additives on insulin absorption from intratracheally administered dry powders in rats. Int. J. Pharm. 2001, 220, 101–110. [Google Scholar] [CrossRef]
- Salem, L.B.; Bosquillon, C.; Dailey, L.; Delattre, L.; Martin, G.; Evrard, B.; Forbes, B. Sparing methylation of β-cyclodextrin mitigates cytotoxicity and permeability induction in respiratory epithelial cell layers in vitro. J. Control. Release 2009, 136, 110–116. [Google Scholar] [CrossRef]
- Shen, Z.-C.; Cheng, Y.; Zhang, Q.; Wei, S.-L.; Li, R.-C.; Wang, K. Lanthanides enhance pulmonary absorption of insulin. Boil. Trace Element Res. 2000, 75, 215–225. [Google Scholar] [CrossRef]
- Morimoto, K.; Uehara, Y.; Iwanaga, K.; Kakemi, M. Effects of sodium glycocholate and protease inhibitors on permeability of trh and insulin across rabbit trachea. Pharm. Acta Helv. 2000, 74, 411–415. [Google Scholar] [CrossRef]
- Johansson, F.; Hjertberg, E.; Eirefelt, S.; Tronde, A.; Bengtsson, U.H. Mechanisms for absorption enhancement of inhaled insulin by sodium taurocholate. Eur. J. Pharm. Sci. 2002, 17, 63–71. [Google Scholar] [CrossRef]
- Yang, T.; Mustafa, F.; Bai, S.; Ahsan, F. Pulmonary delivery of low molecular weight heparins. Pharm. Res. 2004, 21, 2009–2016. [Google Scholar] [CrossRef]
- Martin, G.P.; El-Hariri, L.M.; Marriott, C. Bile salt-and lysophosphatidylcholine-induced membrane damage in human erythrocytes. J. Pharm. Pharmacol. 1992, 44, 646–650. [Google Scholar] [CrossRef]
- Gibaldi, M.; Feldman, S. Mechanisms of surfactant effects on drug absorption. J. Pharm. Sci. 1970, 59, 579–589. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A.; Hornof, M.; Zoidl, T. Thiolated polymers—Thiomers: Synthesis and in vitro evaluation of chitosan–2-iminothiolane conjugates. Int. J. Pharm. 2003, 260, 229–237. [Google Scholar] [CrossRef]
- Bechgaard, E.; Gizurarson, S.; Hjortkjær, R.K.; Sørensen, A.R. Intranasal administration of insulin to rabbits using glycofurol as an absorption promoter. Int. J. Pharm. 1996, 128, 287–289. [Google Scholar] [CrossRef]
- Gizurarson, S.; Marriott, C.; Martin, G.P.; Bechgaard, E. The influence of insulin and some excipients used in nasal insulin preparations on mucociliary clearance. Int. J. Pharm. 1990, 65, 243–247. [Google Scholar] [CrossRef]
- Sørensen, A.; Drejer, K.; Engesgaard, A.; Guldhammer, B.; Hansen, P.; Hjortekjaer, R.; Mygind, N. Rabbit model for studies of nasal administration of insulin and glucagon. Diabet. Res. Clin. Pract. Suppl. 1988, 1, S165. [Google Scholar]
- Bagger, M.A.; Nielsen, H.W.; Bechgaard, E. Nasal bioavailability of peptide T in rabbits: Absorption enhancement by sodium glycocholate and glycofurol. Eur. J. Pharm. Sci. 2001, 14, 69–74. [Google Scholar] [CrossRef]
- Shinichiro, H.; Takatsuka, Y.; Hiroyuki, M. Mechanisms for the enhancement of the nasal absorption of insulin by surfactants. Int. J. Pharm. 1981, 9, 173–184. [Google Scholar] [CrossRef]
- Pontiroli, A.; Alberetto, M.; Pajetta, E.; Calderara, A.; Pozza, G. Human insulin plus sodium glycocholate in a nasal spray formulation: Improved bioavailability and effectiveness in normal subjects. Diabete Metab. 1987, 13, 441–443. [Google Scholar]
- Pontiroli, A.; Alberetto, M.; Calderara, A.; Pajetta, E.; Pozza, G. Nasal administration of glucagon and human calcitonin to healthy subjects: A comparison of powders and spray solutions and of different enhancing agents. Eur. J. Clin. Pharmacol. 1989, 37, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Aungst, B.J.; Rogers, N.J.; Shefter, E. Comparison of nasal, rectal, buccal, sublingual and intramuscular insulin efficacy and the effects of a bile salt absorption promoter. J. Pharmacol. Exp. Ther. 1988, 244, 23–27. [Google Scholar] [PubMed]
- Lee, V.H. Mucosal penetration enhancers for facilitation of peptide and protein drug absorption. Crit. Rev. Ther. Drug Carr. Syst. 1991, 8, 91–192. [Google Scholar]
- Illum, L. Nasal drug delivery: New developments and strategies. Drug Discov. Today 2002, 7, 1184–1189. [Google Scholar] [CrossRef]
- Illum, L.; Farraj, N.F.; Davis, S.S. Chitosan as a novel nasal delivery system for peptide drugs. Pharm. Res. 1994, 11, 1186–1189. [Google Scholar] [CrossRef] [PubMed]
- Casettari, L.; Illum, L. Chitosan in nasal delivery systems for therapeutic drugs. J. Control. Release 2014, 190, 189–200. [Google Scholar] [CrossRef]
- Kotze, A.; Luessen, H.; De Boer, A.; Verhoef, J.; Junginger, H. Chitosan for enhanced intestinal permeability: Prospects for derivatives soluble in neutral and basic environments. Eur. J. Pharm. Sci. 1999, 7, 145–151. [Google Scholar] [CrossRef]
- Farraj, N.; Johansen, B.; Davis, S.; Illum, L. Nasal administration of insulin using bioadhesive microspheres as a delivery system. J. Control. Release 1990, 13, 253–261. [Google Scholar] [CrossRef]
- Illum, L. Chitosan and its use as a pharmaceutical excipient. Pharm. Res. 1998, 15, 1326–1331. [Google Scholar] [CrossRef]
- Illum, L.; Fisher, A.; Jabbal-Gill, I.; Davis, S. Bioadhesive starch microspheres and absorption enhancing agents act synergistically to enhance the nasal absorption of polypeptides. Int. J. Pharm. 2001, 222, 109–119. [Google Scholar] [CrossRef]
- Björk, E.; Edman, P. Characterization of degradable starch microspheres as a nasal delivery system for drugs. Int. J. Pharm. 1990, 62, 187–192. [Google Scholar] [CrossRef]
- Sallee, V.L.; Dietschy, J.M. Determinants of intestinal mucosal uptake of short-and medium-chain fatty acids and alcohols. J. Lipid Res. 1973, 14, 475–484. [Google Scholar]
- Westergaard, H.; Dietschy, J.M. The mechanism whereby bile acid micelles increase the rate of fatty acid and cholesterol uptake into the intestinal mucosal cell. J. Clin. Investig. 1976, 58, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.A. Intestinal transport of bile acids. Am. J. Physiol.-Gastrointest. Liver Physiol. 1981, 241, G83–G92. [Google Scholar] [CrossRef] [PubMed]
- Jalali, A.; Moghimipour, E.; Akhgari, A. Enhancing effect of bile salts on gastrointestinal absorption of insulin. Trop. J. Pharm. Res. 2014, 13, 1797–1802. [Google Scholar] [CrossRef]
- Hersey, S.; Jackson, R. Effect of bile salts on nasal permeability in vitro. J. Pharm. Sci. 1987, 76, 876–879. [Google Scholar] [CrossRef]
- Scott-Moncrieff, J.C.; Shao, Z.; Mitra, A.K. Enhancement of intestinal insulin absorption by bile salt–fatty acid mixed micelles in dogs. J. Pharm. Sci. 1994, 83, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Ali, B.; Amin, S.; Ahmad, J.; Ali, A.; Ali, M.; Mir, S.R. Bioavailability enhancement studies of amoxicillin with nigella. Indian J. Med Res. 2012, 135, 555. [Google Scholar] [PubMed]
- Sinha, V.; Kaur, M.P. Permeation enhancers for transdermal drug delivery. Drug Dev. Ind. Pharm. 2000, 26, 1131–1140. [Google Scholar] [CrossRef]
- Al-Jenoobi, F.; Al-Suwayeh, S.; Muzaffar, I.; Alam, M.A.; Al-Kharfy, K.M.; Korashy, H.M.; Al-Mohizea, A.M.; Ahad, A.; Raish, M. Effects of nigella sativa and lepidium sativum on cyclosporine pharmacokinetics. BioMed Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Bedada, S.K.; Appani, R.; Boga, P.K. Capsaicin pretreatment enhanced the bioavailability of fexofenadine in rats by p-glycoprotein modulation: In vitro, in situ and in vivo evaluation. Drug Dev. Ind. Pharm. 2017, 43, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Tatiraju, D.V.; Bagade, V.B.; Karambelkar, P.J.; Jadhav, V.M.; Kadam, V. Natural bioenhancers: An overview. J. Pharm. Phytochem. 2013, 2, 55–60. [Google Scholar]
- Survase, S.A.; Kagliwal, L.D.; Annapure, U.S.; Singhal, R.S. Cyclosporin A—A review on fermentative production, downstream processing and pharmacological applications. Biotechnol. Adv. 2011, 29, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Thanou, M.; Verhoef, J.; Junginger, H. Oral drug absorption enhancement by chitosan and its derivatives. Adv. Drug Deliv. Rev. 2001, 52, 117–126. [Google Scholar] [CrossRef]
- Prego, C.; Torres, D.; Alonso, M.J. The potential of chitosan for the oral administration of peptides. Expert Opin. Drug Deliv. 2005, 2, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Lueßen, H.L.; de Leeuw, B.J.; Langemeÿer, M.W.; de Boer, A.B.G.; Verhoef, J.C.; Junginger, H.E. Mucoadhesive polymers in peroral peptide drug delivery. Vi. Carbomer and chitosan improve the intestinal absorption of the peptide drug buserelin in vivo. Pharm. Res. 1996, 13, 1668–1672. [Google Scholar] [CrossRef]
- Schipper, N.G.; Vårum, K.M.; Artursson, P. Chitosans as absorption enhancers for poorly absorbable drugs. 1: Influence of molecular weight and degree of acetylation on drug transport across human intestinal epithelial (caco-2) cells. Pharm. Res. 1996, 13, 1686–1692. [Google Scholar] [CrossRef]
- Kotzé, A.F.; Lueßen, H.L.; de Leeuw, B.J.; Verhoef, J.C.; Junginger, H.E. Comparison of the effect of different chitosan salts and n-trimethyl chitosan chloride on the permeability of intestinal epithelial cells (caco-2). J. Control. Release 1998, 51, 35–46. [Google Scholar] [CrossRef]
- Borchard, G.; Lueβen, H.L.; de Boer, A.G.; Verhoef, J.C.; Lehr, C.-M.; Junginger, H.E. The potential of mucoadhesive polymers in enhancing intestinal peptide drug absorption. Iii: Effects of chitosan-glutamate and carbomer on epithelial tight junctions in vitro. J. Control. Release 1996, 39, 131–138. [Google Scholar] [CrossRef]
- Smith, J.; Wood, E.; Dornish, M. Effect of chitosan on epithelial cell tight junctions. Pharm. Res. 2004, 21, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Heydt, M.S.; Amasheh, M.; Stein, C.; Fromm, M.; Amasheh, S. Analysis of absorption enhancers in epithelial cell models. Ann. N. Y. Acad. Sci. 2012, 1258, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Werle, M.; Takeuchi, H.; Bernkop-Schnürch, A. Modified chitosans for oral drug delivery. J. Pharm. Sci. 2009, 98, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Cavaleri, F.; Jia, W. The true nature of curcumin’s polypharmacology. J. Prev. Med. 2017, 2, 5. [Google Scholar] [CrossRef]
- Bahramsoltani, R.; Rahimi, R.; Farzaei, M.H. Pharmacokinetic interactions of curcuminoids with conventional drugs: A review. J. Ethnopharmacol. 2017, 209, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Lee, W.; Choi, J. Effects of curcumin on the pharmacokinetics of tamoxifen and its active metabolite, 4-hydroxytamoxifen, in rats: Possible role of cyp3a4 and p-glycoprotein inhibition by curcumin. Die Pharm. Int. J. Pharm. Sci. 2012, 67, 124–130. [Google Scholar]
- Abo-El-Sooud, K.; Samar, M.; Fahmy, M. Curcumin ameliorates the absolute and relative bioavailabilities of marbofloxacin after oral administrations in broiler chickens. Wulfenia 2017, 24, 284–297. [Google Scholar]
- Ampasavate, C.; Sotanaphun, U.; Phattanawasin, P.; Piyapolrungroj, N. Effects of curcuma spp. On p-glycoprotein function. Phytomedicine 2010, 17, 506–512. [Google Scholar] [CrossRef]
- Neerati, P.; Bedada, S.K. Effect of diosmin on the intestinal absorption and pharmacokinetics of fexofenadine in rats. Pharmacol. Rep. 2015, 67, 339–344. [Google Scholar] [CrossRef]
- Wang, J.-B.; Zhao, H.-P.; Zhao, Y.-L.; Jin, C.; Liu, D.-J.; Kong, W.-J.; Fang, F.; Zhang, L.; Wang, H.-J.; Xiao, X.-H. Hepatotoxicity or hepatoprotection? Pattern recognition for the paradoxical effect of the chinese herb rheum palmatum l. In treating rat liver injury. PLoS ONE 2011, 6, e24498. [Google Scholar] [CrossRef]
- Lee, M.-H.; Kao, L.; Lin, C.-C. Comparison of the antioxidant and transmembrane permeative activities of the different polygonum cuspidatum extracts in phospholipid-based microemulsions. J. Agric. Food Chem. 2011, 59, 9135–9141. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, R.; Wang, W.; Mao, X.; Yu, J. Lipid regulation effects of polygoni multiflori radix, its processed products and its major substances on steatosis human liver cell line l02. J. Ethnopharmacol. 2012, 139, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-C.; Lim, M.-Y.; Lee, H.-S. Emodin isolated from cassia obtusifolia (leguminosae) seed shows larvicidal activity against three mosquito species. J. Agric. Food Chem. 2003, 51, 7629–7631. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, S.; Ullah, M.; Hadi, S. DNA Degradation by Aqueous Extract of Aloe Vera in the Presence Of Copper Ions; CSIR: New Delhi, India, 2010. [Google Scholar]
- Yang, T.; Kong, B.; Kuang, Y.; Cheng, L.; Gu, J.; Zhang, J.; Shu, H.; Yu, S.; Yang, X.; Cheng, J. Emodin plays an interventional role in epileptic rats via multidrug resistance gene 1 (mdr1). Int. J. Clin. Exp. Pathol. 2015, 8, 3418. [Google Scholar] [PubMed]
- Ko, J.-C.; Su, Y.-J.; Lin, S.-T.; Jhan, J.-Y.; Ciou, S.-C.; Cheng, C.-M.; Chiu, Y.-F.; Kuo, Y.-H.; Tsai, M.-S.; Lin, Y.-W. Emodin enhances cisplatin-induced cytotoxicity via down-regulation of ercc1 and inactivation of erk1/2. Lung Cancer 2010, 69, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Lu, J.; Huang, M.; Li, Y.; Chen, M.; Wu, G.; Gong, J.; Zhong, Z.; Xu, Z.; Dang, Y. Anti-cancer natural products isolated from chinese medicinal herbs. Chin. Med. 2011, 6, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Li, W.; Shi, H.; Xie, X.; Li, L.; Tang, H.; Wu, M.; Kong, Y.; Yang, L.; Gao, J. Synergistic effects of curcumin with emodin against the proliferation and invasion of breast cancer cells through upregulation of mir-34a. Mol. Cell. Biochem. 2013, 382, 103–111. [Google Scholar] [CrossRef]
- Min, H.; Niu, M.; Zhang, W.; Yan, J.; Li, J.; Tan, X.; Li, B.; Su, M.; Di, B.; Yan, F. Emodin reverses leukemia multidrug resistance by competitive inhibition and downregulation of p-glycoprotein. PLoS ONE 2017, 12, e0187971. [Google Scholar] [CrossRef]
- Choi, R.J.; Ngoc, T.M.; Bae, K.; Cho, H.-J.; Kim, D.-D.; Chun, J.; Khan, S.; Kim, Y.S. Anti-inflammatory properties of anthraquinones and their relationship with the regulation of p-glycoprotein function and expression. Eur. J. Pharm. Sci. 2013, 48, 272–281. [Google Scholar] [CrossRef]
- Huang, J.; Guo, L.; Tan, R.; Wei, M.; Zhang, J.; Zhao, Y.; Gong, L.; Huang, Z.; Qiu, X. Interactions between emodin and efflux transporters on rat enterocyte by a validated ussing chamber technique. Front. Pharmacol. 2018, 9, 646. [Google Scholar] [CrossRef]
- Mansouri, M.T.; Soltani, M.; Naghizadeh, B.; Farbood, Y.; Mashak, A.; Sarkaki, A. A possible mechanism for the anxiolytic-like effect of gallic acid in the rat elevated plus maze. Pharmacol. Biochem. Behav. 2014, 117, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Stupans, I.; Stretch, G.; Hayball, P. Olive oil phenols inhibit human hepatic microsomal activity. J. Nutr. 2000, 130, 2367–2370. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Yoshida, T.; Hatano, T. Hydrolyzable tannins and related polyphenols. In Fortschritte der Chemie Organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products; Springer: New York, NY, USA, 1995; pp. 1–117. [Google Scholar]
- Chieli, E.; Romiti, N.; Rodeiro, I.; Garrido, G. In vitro modulation of abcb1/p-glycoprotein expression by polyphenols from mangifera indica. Chem. Boil. Interact. 2010, 186, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Basheer, L.; Kerem, Z. Interactions between cyp3a4 and dietary polyphenols. Oxidative Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef]
- Athukuri, B.L.; Neerati, P. Enhanced oral bioavailability of diltiazem by the influence of gallic acid and ellagic acid in male wistar rats: Involvement of cyp3a and p-gp inhibition. Phytother. Res. 2017, 31, 1441–1448. [Google Scholar] [CrossRef]
- Athukuri, B.L.; Neerati, P. Enhanced oral bioavailability of metoprolol with gallic acid and ellagic acid in male wistar rats: Involvement of cyp2d6 inhibition. Drug Metab. Pers. Ther. 2016, 31, 229–234. [Google Scholar] [CrossRef]
- Kesarwani, K.; Gupta, R. Bioavailability enhancers of herbal origin: An overview. Asian Pac. J. Trop. Biomed. 2013, 3, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Bhadoriya, S.S.; Mangal, A.; Madoriya, N.; Dixit, P. Bioavailability and bioactivity enhancement of herbal drugs by “nanotechnology”: A review. J. Curr. Pharm. Res. 2011, 8, 1–7. [Google Scholar]
- Hayeshi, R.; Masimirembwa, C.; Mukanganyama, S.; Ungell, A.-L.B. The potential inhibitory effect of antiparasitic drugs and natural products on p-glycoprotein mediated efflux. Eur. J. Pharm. Sci. 2006, 29, 70–81. [Google Scholar] [CrossRef]
- Limtrakul, P.; Khantamat, O.; Pintha, K. Inhibition of p-glycoprotein function and expression by kaempferol and quercetin. J. Chemother. 2005, 17, 86–95. [Google Scholar] [CrossRef]
- Bobrowska-Hägerstrand, M.; Wróbel, A.; Rychlik, B.; Bartosz, G.; Söderström, T.; Shirataki, Y.; Motohashi, N.; Molnár, J.; Michalak, K.; Hägerstrand, H. Monitoring of mrp-like activity in human erythrocytes: Inhibitory effect of isoflavones. Blood Cells Mol. Dis. 2001, 27, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Dube, A.; Nicolazzo, J.A.; Larson, I. Chitosan nanoparticles enhance the plasma exposure of (−)-epigallocatechin gallate in mice through an enhancement in intestinal stability. Eur. J. Pharm. Sci. 2011, 44, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Otero, J.A.; Barrera, B.; Prieto, J.G.; Merino, G.; Alvarez, A.I. Inhibition of abcg2/bcrp transporter by soy isoflavones genistein and daidzein: Effect on plasma and milk levels of danofloxacin in sheep. Vet. J. 2013, 196, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, C.; Zhou, T.; Chen, G. Genistein induction of human sulfotransferases in hepg2 and caco-2 cells. Basic Clin. Pharmacol. Toxicol. 2008, 103, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Eaton, E.A.; Walle, U.K.; Lewis, A.J.; Hudson, T.; Wilson, A.A.; Walle, T. Flavonoids, potent inhibitors of the human p-form phenolsulfotransferase. Potential role in drug metabolism and chemoprevention. Drug Metab. Dispos. 1996, 24, 232–237. [Google Scholar] [PubMed]
- Reddy, S.A.; Sen, S.; Chakraborty, R.; Parameshappa, B. Effect of gokhru plant extract on intestinal absorption of aspirin using everted sac technique. Int. J. Pharm. Boil. Arch. 2011, 2, 549–553. [Google Scholar]
- Bailey, D.G.; Malcolm, J.; Arnold, O.; Spence, J.D. Grapefruit juice–drug interactions. Br. J. Clin. Pharmacol. 1998, 46, 101–110. [Google Scholar] [CrossRef]
- Mouly, S.; Lloret-Linares, C.; Sellier, P.-O.; Sene, D.; Bergmann, J.-F. Is the clinical relevance of drug-food and drug-herb interactions limited to grapefruit juice and saint-john’s wort? Pharmacol. Res. 2017, 118, 82–92. [Google Scholar] [CrossRef]
- Bailey, D.; Spence, J.; Munoz, C.; Arnold, J. Interaction of citrus juices with felodipine and nifedipine. Lancet 1991, 337, 268–269. [Google Scholar] [CrossRef]
- Mertens-Talcott, S.; Zadezensky, I.; Castro, W.; Derendorf, H.; Butterweck, V. Grapefruit-drug interactions: Can interactions with drugs be avoided? J. Clin. Pharmacol. 2006, 46, 1390–1416. [Google Scholar] [CrossRef]
- Ahmed, I.S.; Hassan, M.A.; Kondo, T. Effect of lyophilized grapefruit juice on p-glycoprotein-mediated drug transport in-vitro and in-vivo. Drug Dev. Ind. Pharm. 2015, 41, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Butterweck, V.; Zdrojewski, I.; Galloway, C.; Frye, R.; Derendorf, H. Toxicological and pharmacokinetic evaluation of concomitant intake of grapefruit juice and simvastatin in rats after repeated treatment over 28 days. Planta Med. 2009, 75, 1196. [Google Scholar] [CrossRef] [PubMed]
- Shirasaka, Y.; Shichiri, M.; Mori, T.; Nakanishi, T.; Tamai, I. Major active components in grapefruit, orange, and apple juices responsible for oatp2b1-mediated drug interactions. J. Pharm. Sci. 2013, 102, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Spahn-Langguth, H.; Langguth, P. Grapefruit juice enhances intestinal absorption of the p-glycoprotein substrate talinolol. Eur. J. Pharm. Sci. 2001, 12, 361–367. [Google Scholar] [CrossRef]
- Tian, R.; Koyabu, N.; Takanaga, H.; Matsuo, H.; Ohtani, H.; Sawada, Y. Effects of grapefruit juice and orange juice on the intestinal efflux of p-glycoprotein substrates. Pharm. Res. 2002, 19, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Lown, K.S.; Bailey, D.G.; Fontana, R.J.; Janardan, S.K.; Adair, C.H.; Fortlage, L.A.; Brown, M.B.; Guo, W.; Watkins, P.B. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal cyp3a protein expression. J. Clin. Investig. 1997, 99, 2545–2553. [Google Scholar] [CrossRef]
- Dresser, G.K.; Kim, R.B.; Bailey, D.G. Effect of grapefruit juice volume on the reduction of fexofenadine bioavailability: Possible role of organic anion transporting polypeptides. Clin. Pharmacol. Ther. 2005, 77, 170–177. [Google Scholar] [CrossRef]
- Tamai, I.; Nakanishi, T. Oatp transporter-mediated drug absorption and interaction. Curr. Opin. Pharmacol. 2013, 13, 859–863. [Google Scholar] [CrossRef]
- Khanuja, S.; Arya, J.; Srivastava, S.; Shasany, A.; Kumar, T.S.; Darokar, M.; Kumar, S. Antibiotic Pharmaceutical Composition with Lysergol as Bio-Enhancer and Method of Treatment. U.S. Patent 11/395,527, 15 March 2007. [Google Scholar]
- Alexander, A.; Qureshi, A.; Kumari, L.; Vaishnav, P.; Sharma, M.; Saraf, S.; Saraf, S. Role of herbal bioactives as a potential bioavailability enhancer for active pharmaceutical ingredients. Fitoterapia 2014, 97, 1–14. [Google Scholar] [CrossRef]
- Paine, M.F.; Criss, A.B.; Watkins, P.B. Two major grapefruit juice components differ in intestinal cyp3a4 inhibition kinetic and binding properties. Drug Metab. Dispos. 2004. [Google Scholar] [CrossRef]
- Hochman, J.H.; Fix, J.A.; LeCluyse, E.L. In vitro and in vivo analysis of the mechanism of absorption enhancement by palmitoylcarnitine. J. Pharmacol. Exp. Ther. 1994, 269, 813–822. [Google Scholar] [PubMed]
- LeCluyse, E.L.; Appel, L.E.; Sutton, S.C. Relationship between drug absorption enhancing activity and membrane perturbing effects of acylcarnitines. Pharm. Res. 1991, 8, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Duizer, E.; Van Der Wulp, C.; Versantvoort, C.H.; Groten, J.P. Absorption enhancement, structural changes in tight junctions and cytotoxicity caused by palmitoyl carnitine in caco-2 and iec-18 cells. J. Pharmacol. Exp. Ther. 1998, 287, 395–402. [Google Scholar] [PubMed]
- Bhardwaj, R.K.; Glaeser, H.; Becquemont, L.; Klotz, U.; Gupta, S.K.; Fromm, M.F. Piperine, a major constituent of black pepper, inhibits human p-glycoprotein and cyp3a4. J. Pharmacol. Exp. Ther. 2002, 302, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Fazal, H.; Abbasi, B.H.; Farooq, S.; Ali, M.; Khan, M.A. Biological role of piper nigrum l.(black pepper): A review. Asian Pac. J. Trop. Biomed. 2012, 2, S1945–S1953. [Google Scholar] [CrossRef]
- Riedmaier, A.E.; Nies, A.T.; Schaeffeler, E.; Schwab, M. Organic anion transporters and their implications in pharmacotherapy. Pharmacol. Rev. 2012, 64, 421–449. [Google Scholar] [CrossRef]
- Fukuhara, K.; Ikawa, K.; Morikawa, N.; Kumagai, K. Population pharmacokinetics of high-dose methotrexate in japanese adult patients with malignancies: A concurrent analysis of the serum and urine concentration data. J. Clin. Pharm. Ther. 2008, 33, 677–684. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Zhang, J.; Yu, X.; Wang, X.; Zhao, L. Effects of resveratrol on p-glycoprotein and cytochrome p450 3a in vitro and on pharmacokinetics of oral saquinavir in rats. Drug Des. Dev. Ther. 2016, 10, 3699. [Google Scholar] [CrossRef]
- Sharma, P.; Varma, M.V.; Chawla, H.P.; Panchagnula, R. In situ and in vivo efficacy of peroral absorption enhancers in rats and correlation to in vitro mechanistic studies. IL Farmaco 2005, 60, 874–883. [Google Scholar] [CrossRef]
- Fasano, A.; Fiorentini, C.; Donelli, G.; Uzzau, S.; Kaper, J.B.; Margaretten, K.; Ding, X.; Guandalini, S.; Comstock, L.; Goldblum, S.E. Zonula occludens toxin modulates tight junctions through protein kinase c-dependent actin reorganization, in vitro. J. Clin. Investig. 1995, 96, 710–720. [Google Scholar] [CrossRef]
- Yang, T.; Hussain, A.; Paulson, J.; Abbruscato, T.J.; Ahsan, F. Cyclodextrins in nasal delivery of low-molecular-weight heparins: In vivo and in vitro studies. Pharm. Res. 2004, 21, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Yao, H.; Lin, H.; Lu, J.; Li, R.; Wang, K. The events relating to lanthanide ions enhanced permeability of human erythrocyte membrane: Binding, conformational change, phase transition, perforation and ion transport. Chem. Boil. Interact. 1999, 121, 267–289. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, M.; Li, R.; Wang, C.; Bai, C.; Wang, K. Gadolinium induces domain and pore formation of human erythrocyte membrane: An atomic force microscopic study. Biochim. Biophys. Acta 1999, 1421, 249–260. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Route of Administration | Bioenhancer (Class) | Biological Source | Mechanism(s) of Action | Study Design Model | Research Compound | Reference(s) |
|---|---|---|---|---|---|---|
| Buccal | Aloe vera (gel, whole leaf) | Plant (Aloe vera) | Intercellular modulation | In vitro (Franz diffusion cells) | Didanosine: Antiviral reverse transcriptase inhibitor | [20] |
| Buccal | Chitosan (Biopolymer) | Deacetylated chitin from crustaceans and fungi | Mucoadhesion; changes in lipid organization and loosening of intercellular filaments | In vitro (T146 cells 1) | FITC–dextran: Hydrophilic polysaccharide | [21] |
| Buccal | Chitosan (Biopolymer) | Deacetylated chitin from crustaceans and fungi | Mucoadhesion; mucosal membrane modulation | Ex vivo (porcine buccal mucosa) | Hydrocortisone: Corticosteroid TGF-beta: Cytokine polypeptide | [22] |
| Buccal | Chitosan–TBA (Thiolated polymer) | Deacetylated chitin from crustaceans and fungi | Mucoadhesion; mucosal membrane modulation | Ex vivo (porcine buccal mucosa); in vivo (pig) | PACAP: Pituitary Adenylate Cyclase-activating Peptide | [23,24] |
| Buccal | Cod-liver oil extract (Fatty acid) | Animal (Cod fish) | No mechanism specified | Ex vivo (hamster cheek pouch) | Ergotamine tartrate: Ergopeptine alkaloid | [25] |
| Buccal | Menthol (Alcohol) | Plant (Corn mint, peppermint, or other mint oils) | No mechanism specified | Ex vivo (porcine buccal mucosa) | Dideoxycytidine: Nucleoside analog reverse transcriptase inhibitor (NRTI) | [26] |
| Buccal | Oleic acid, eicosapentaenoic acid, docosahexaenoic acid (Fatty acids) | Animal (Cod fish) | No mechanism specified | In vitro (membraneless dissolution test), in vivo (rat) | Insulin: Peptide hormone | [27] |
| Buccal | Sodium glycodeoxycholate (Bile salt) | Intestinal bacterial by-product | No mechanism specified | Ex vivo (porcine buccal mucosa) | Dideoxycytidine: Nucleoside analog reverse transcriptase inhibitor (NRTI) | [28] |
| Buccal | TMC (Cationic polymers) | Chemically modified chitosan (crustaceans, fungi) | Mucoadhesion; mucosal membrane modulation | Ex vivo (porcine buccal mucosa) | FD4: Hydrophilic polysaccharide | [29] |
| Nasal | Chitosan (Biopolymer) | Chemically modified chitosan (crustaceans, fungi) | Tight junction modulation | In vivo (sheep) | sCT: Endogenous polypeptide hormone | [30] |
| Nasal | Chitosan (Biopolymer) | Deacetylated chitin from crustaceans and fungi | Increased mucoadhesion; tight junction modulation | In vivo (sheep, human) | Morphine: Opium alkaloid | [31] |
| Nasal | Chitosan–TBA (Thiolated polymer) | Deacetylated chitin from crustaceans and fungi | Increased mucoadhesion; tight junction modulation | In vivo (rat) | Insulin: Peptide hormone | [32] |
| Nasal | TMC (Cationic polymers) | Chemically modified chitosan (crustaceans and fungi) | Increased mucoadhesion; tight junction modulation | In vivo (rat) | Mannitol: Sugar alcohol | [33] |
| Oral | (-)-Epicatechin (Flavonoid) | Plant (woody plants) | Metabolism (glucuronidation) inhibition | Ex vivo (rat small intestine) | Alpha-naphtol: Organic fluorescent compound | [34] |
| Oral | Aloe vera (gel and whole leaf) | Plant (Aloe vera) | Tight junction modulation | Ex vivo (rat intestinal tissue) | Atenolol: Beta-receptor activity compound | [35] |
| Oral | Aloe vera (gel and whole leaf) | Plant (Aloe vera) | Tight junction modulation | In vitro (Caco-2 cells 2) | Insulin: Peptide hormone | [36] |
| Oral | Aloe vera (juice) | Plant (Aloe vera) | Local mucosal tissue modulation | In vivo (human) | Vitamin C and E: Ascorbic acid, tocopherols, tocotrienols. | [37] |
| Oral | Aloe vera (gel polysaccharides) | Plant (Aloe vera) | Metabolism inhibition; tight junction modulation | In vitro (Caco-2, LS180 cells 3), In vivo (rat) | Indinavir: Antiviral protease inhibitor | [38] |
| Oral | BHCl (Flavonoid) | Acidification of betaine Plant (beetroot: Beta vulgaris) | Metabolism enhancement (transient re-acidification of gastric pH) | In vivo (human) | Dasatinib: Protein kinase inhibitor | [39] |
| Oral | Caraway (Flavonoid) | Plant (meridian fennel/Persian cumin: Carum carvi) | Local mucosal tissue modulation | In vivo (human) | Rifampicin: Semisynthetic rifamycin derivative, Isoniazid: Isonicotinic acid derivative, pyrazinamide: nicotinamide pyrazine analogue | [40] |
| Oral | Chitosan (Biopolymer) | Deacetylated chitin from crustaceans and fungi | Tight junction modulation | In vitro (HT-29 clone B6 cells 4) | Heparin: Anticoagulant | [41] |
| Oral | Chitosan (Biopolymer) | Deacetylated chitin from crustaceans and fungi | Tight junction modulation | In vitro (Caco-2 cells 2) | Chitosan– (Lissamine–rhodamine labelled) | [42] |
| Oral | Chitosan–TBA (Thiolated polymer) | Deacetylated chitin from crustaceans and fungi | Mucoadhesion; tight junction modulation | Ex vivo (guinea pig small intestinal mucosa) | Cefadroxil: Cephalosporin | [43] |
| Oral | Chitosan–TBA (Thiolated polymer) | Deacetylated chitin from crustaceans and fungi | Mucoadhesion; tight junction modulation | In vivo (rat) | Insulin: Peptide hormone | [44] |
| Oral | Curcumin (Flavonoid) | Plant (turmeric: Curcuma longa) | Metabolism (UDP-glucuronyl transferase) inhibition | In vitro (rat microsomes) | Mycohenolic acid: Immunosuppressant | [45] |
| Oral | Curcumin (Flavonoid) | Plant (turmeric: Curcuma longa) | Efflux transporter inhibition; metabolism inhibition | In vivo (rabbit) | Norfloxacin: Fluoroquinolone | [46] |
| Oral | Curcumin (Flavonoid) | Plant (turmeric: Curcuma longa) | Metabolism (CYP3A4) inhibition | In vitro (human liver microsomes) | Midazolam: Benzodiazepine | [47] |
| Oral | Curcumin (Flavonoid) | Plant (turmeric: Curcuma longa) | Efflux transporter (P-gp) inhibition; metabolism (CYP3A4) inhibition | In vivo (rat) | Midazolam: Benzodiazepine | [48] |
| Oral | Cyclosporine A (Immunosuppressant) | Fungi (Tolypocladium inflatum Gams) | Efflux transporter (P-gp) inhibition | In vivo (rat, dog) | Clopidogrel: Platelet aggregation inhibitor | [49] |
| Oral | Diosmin (Flavonoid) | Plant (citrus fruits) | Efflux transporter (P-gp) inhibition | In vitro (Caco-2 cells 2) | Digoxin: Digitalis glycoside | [50] |
| Oral | Emodin (Anthraquinone derivative) | Plant (senna: Cassia angustifolia, Aloe vera (syn Aloe barbadensis), rhubarb: Rheum officinale) | Efflux transporter (P-gp) inhibition | In vitro (MDR1-MDCKII cells 6, Caco-2 cells2) | Digoxin: Digitalis glycoside | [51] |
| Oral | Fulvic acid (Organic acid) | Plant (decomposed material) | Metabolism enhancement (enhanced drug water solubility) | In vivo (rat) | Glibenclamide: Sulfonylurea antidiabetic Insulin: Peptide hormone Pentazocin: Opioid analgesic | [52] |
| Oral | Gallic acid ester (Organic acid) | Plant (gallnuts, sumac, witch hazel, tea leaves, oak bark) | Metabolism (CYP3A) inhibition | In vitro (human liver microsomes) | Nifedipine: Calcium channel blocker | [53] |
| Oral | Genistein (Flavonoid) | Plant (soyabean: Glycine max, kudzu: Pueraria lobata) | Efflux transporter (MRP) inhibition | In vitro (HT-29 cells4), In vivo (rat) | Epigalllocatechin-3-gallate (EGCG): Phenolic antioxidant | [54] |
| Oral | Genistein (Flavonoid) | Plant (soyabean: Glycine max, kudzu: Pueraria lobata) | Efflux transporter (P-gp, BCRP, MRP2) inhibition; metabolism (CYP3A4) inhibition | In vivo (rat) | Paclitaxel: Tetracyclic diterpenoid | [55] |
| Oral | Gokhru extract (Herbal) | Plant (Tribulus: Tribulus terrestris) | Local mucosal tissue modulation | In vitro (goat everted sac) | Metformin: Biguanide | [56] |
| Oral | Gokhru extract (Herbal) | Plant (Tribulus: Tribulus terrestris) | Local mucosal tissue modulation | In vitro (chicken everted intestine) | Metformin: Biguanide | [57] |
| Oral | Grapefruit juice (Citrus fruit) | Plant (grapefruit: Citrus paradisi) | Efflux transporter (P-gp, MRP2); metabolism (CYP3A4) inhibition; renal uptake transporter (OATP) inhibition | Various | Various | [58] |
| Oral | LSC (Chitosan derivative) | Modified chitosan (crustaceans and fungi) | Increased mucoadhesion; tight junction modulation | In vitro (Caco-2 cells 2), In vivo (rat), Ex vivo (rat intestine) | Insulin: Peptide hormone | [59] |
| Oral | Lycopene (Carotenoid) | Plant (red fruits and vegetables) | Dual carotenoid/LDL receptor mechanism for targeted hepatic delivery | In vivo (human) | Simvastatin: HMG–CoA reductase inhibitor | [60] |
| Oral | Lysergol (Alkaloid) | Plant (morning glory plant: Ipomoea spp.) | Metabolism inhibition | In vivo (rat) | Berberine: Benzylisoquinoline alkaloid | [61] |
| Oral | Lysergol (Alkaloid) | Plant (morning glory plant: Ipomoea spp.) | Efflux transporter (BCRP) inhibition; metabolism inhibition | In vitro (rat liver microsomes) | Curcumin: Zingiberaceae Sulfasalazine: Aminosalicylic agent | [62] |
| Oral | Moringa oleifera pods (Traditional herbal medicine) | Plant (Moringa oleifera) | Metabolism (CYP450) inhibition | In vivo (mice) | Rifampicin: Semisynthetic rifamycin derivative | [63] |
| Oral | Naringin (Flavonoid glycoside) | Plant (grapefruit, apple, onion, tea) | Efflux transporter (P-gp) inhibition; metabolism inhibition | In vivo (rat) | Diltiazem: Benzothiazepine derivates | [64] |
| Oral | Naringin (Flavonoid glycoside) | Plant (grapefruit, apple, onion, tea) | Metabolism (CYP3A4) inhibition | In vivo (rat) | Tamoxifen: selective estrogen receptor modulator (SERM) | [65] |
| Oral | Naringin (Flavonoid glycoside) | Plant (grapefruit, apple, onion, tea) | Efflux transporter (P-gp) inhibition; metabolism (CYP3A4) inhibition | In vivo (rat) | Paclitaxel: Tetracyclic diterpenoid | [66] |
| Oral | Naringin (Flavonoid glycoside) | Plant (grapefruit, apple, onion, tea) | Efflux transporter (P-gp) inhibition; metabolism (CYP3A4) inhibition | Ex vivo (rat everted gut sac) | Clopidogrel: Platelet aggregation inhibitor | [67] |
| Oral | Naringin (Flavonoid glycoside) | Plant (grapefruit, apple, onion, tea) | Metabolism (CYP3A4) inhibition | In vivo (rabbit) | Verapamil: Calcium channel blocker | [68] |
| Oral | Palmitoyl carnitine chloride (Chelating agents) | Esterification of carnitinePlant/animal (various) | Tight junction modulation | In vitro (Caco-2 cells 2) | Clodronate: Bisphosphonate | [69] |
| Oral | Peppermint oil (Herbal) | Plant (peppermint: Mentha pipertita) | Metabolism (CYP3A) inhibition | Ex vivo (rat intestinal tissue) | Cyclosporine: Immunosuppressant | [70] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Local mucosal tissue modulation; thermogenic activity | In vivo (human) | B-carotene: Terpenoid | [71] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Local mucosal tissue modulation; thermogenic activity | In vivo (human) | Coenzyme Q10: benzoquinone | [72] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Decreased elimination (gastrointestinal transit inhibition; gastric emptying inhibition) | In vivo (rat, mice) | Phenol red: Spheroid | [73] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Metabolism inhibition | In vivo (human) | Propanol: Beta-receptor activity compound, theophylline: methylxanthine | [74] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Metabolism (CYP450) inhibition | In vivo (rat) | Nimesulide: Non-steroidal anti-inflammatory | [75] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Efflux transporter (P-gp) inhibition | In vivo (rat) | Fexofenadine: Terfenadine metabolite | [76] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Metabolism inhibition | In vivo (mice) | Resveratrol: Phytoalexin | [77] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Metabolism inhibition | In vivo (human) | Nevirapine: Non-nucleoside reverse transcriptase inhibitor | [78] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Metabolism inhibition | In vivo (mice) | Epigalllocatechin-3-gallate (EGCG): Phenolic antioxidant | [79] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Metabolism inhibition | In vivo (rat) | Pentobarbitone: Barbiturate. | [80] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Metabolism (CYP3A4) inhibition | In vivo (human) | Carbamazepine: Carboxamide derivative | [81] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Metabolism (CYP450) inhibition | In vivo (rat) | Nateglinide: Meglitinide | [82] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Metabolism (hepatic and intestinal glucuronidation) inhibition | In vivo (rat, human) | Curcumin: Zingiberaceae agent | [83] |
| Oral | Piperine (Alkaloid) | Plant (Piper longum and Piper nigrum) | Metabolism inhibition | In vivo (hen) | Oxytetracycline: Bacterial protein synthesis inhibitor | [84] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (P-gp) inhibition | In vivo (rat), Ex vivo (rat and chick everted intestinal sac) | Ranolazine: Piperazine derivative | [85] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (P-gp) inhibition | In vivo (rat), In vitro (Caco-2 cells 2) | Irinotecan: Cytotoxic alkaloid | [86] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (P-gp) inhibition | In vivo (rats), Ex vivo (rat intestinal everted sac) | Valsartan: Angiotensin II receptor antagonist | [87] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Metabolism (CYP3A) inhibition | In vivo (rabbit) | Verapamil: Calcium channel blocker | [88] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (P-gp) inhibition; metabolism (CYP3A) inhibition | In vivo (rabbit) | Dilitiazem: Nondihydropyridine calcium channel blocker | [89] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (P-gp) inhibition; metabolism (CYP3A) inhibition | In vivo (rat) | Doxorubicin: Daunorubicin precursor | [90] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (P-gp) inhibition | In vivo (human) | Fexofenadine: Terfenadine metabolite | [91] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (P-gp) inhibition | In vivo (rat, dog) | Clopidogrel: Platelet aggregation inhibitor | [49] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (P-gp) inhibition; metabolism (CYP3A) inhibition | In vivo (rat) | Etoposide: Podophyllotoxin derivative | [92] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (P-gp) inhibition; metabolism (CYP3A) inhibition | Various | Epigalllocatechin-3-gallate (EGCG): Phenolic antioxidant | [93] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (P-gp) inhibition | In vitro (human MCF-7 ADRr cells 7) | Doxorubicin: Daunorubicin precursor | [94] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Efflux transporter (MRP) inhibition; metabolism (CYP3A) inhibition | In vivo (rat) | Tamoxifen: selective estrogen receptor modulator (SERM) | [95] |
| Oral | Quercetin (Flavonoid) | Plant (citrus fruits, vegetables, leaves, grains) | Metabolism (CYP3A) inhibition | In vivo (rat) | Pioglitazone: Thiazolidinedione | [96] |
| Oral | Quinidine (Class I antiarrhythmic agent) | Chemically modified: stereoisomer of quinine Plant (cinchona tree: Cinchona sp.) | Efflux transporter (P-gp) inhibition | Ex vivo (everted rat gut sac) | Paeoniflorin: Paeonia lactiflora derivative | [97] |
| Oral | Resveratrol (Polyphenolic phytoalexin) | Plant (berries, grape skins, red wine) | Metabolism (CYP2C9, CYP2E1) inhibition | In vivo (human) | Diclofenac: NSAID | [98] |
| Oral | Resveratrol (Polyphenolic phytoalexin) | Plant (berries, grape skins, red wine) | Efflux transporter (P-gp, MRP-2) inhibition; reduced elimination; renal uptake transporter (OAT1, OAT3) inhibition | In vitro (Caco-2 cells 2, mock-MDCK, MDR1-MDCK 6, MRP2-MDCK 6, mock-HEK293, hOAT1-HEK293 8, hOAT3-HEK293 8 cells), Ex vivo (rat everted intestine, rat kidney slices), In vivo (rat) | Methotrexate: Immunosuppressant | [99] |
| Oral | Sinomenine (Alkaloid) | Plant (Sinomenium acutum) | Efflux transporter (P-gp) inhibition | Ex vivo (everted rat gut sac) | Paeoniflorin: Paeonia lactiflora derivative | [97] |
| Oral | Sinomenine (Alkaloid) | Plant (Sinomenium acutum) | Efflux transporter (P-gp) inhibition | In vivo (rat) | Paeoniflorin: Paeonia lactiflora derivative | [100] |
| Oral | Sodium caprate (Fatty acid) | Chemically modified: salification of caproic acid Animal (fats and oils) | Tight junction modulation | In situ (recirculating intestinal perfusion), ex vivo (everted rat gut sacs), in vivo (rat) | Berberine: Antidiabetic plant alkaloid | [101] |
| Oral | Sodium cholate/phospholipid-mixed micelles (Bile salts) | Intestinal bacterial by-product | Mucosal membrane modulation | In vivo (dog) | Silybin, the major active component of silymarin (antihepatotoxic polyphenolic substance isolated from milk thistle plant, Silybum marianum) | [102] |
| Oral | Soybean phosphotidylcholine/sodium deoxycholate (SPC/SDC) (Bile salts) | SPC: plant (soya bean: Glycine max) SDC: chemically modified: salification of deoxycholic acid (metabolic byproduct of intestinal bacteria) | Mucosal membrane modulation | In vivo (dog) | Fenofibrate | [103] |
| Oral | Tamarixetin (metabolite of quercetin) (Flavonoid) | Plant (hogweed/cow parsnip: Heracleum stenopterum) | Metabolism (CYP2C isozyme) inhibition | In vitro (rat liver microsomes), In vivo (rat) | Fluvastatin: HMG CoA reductase inhibitor | [104] |
| Oral | TMC (Cationic polymers) | Modified chitosan (crustaceans, fungi) | Mucoadhesion; tight junction modulation | In vitro (Caco-2 cells 2) | Mannitol: Sugar alcohol PEG 4000: Polyethylene glycol | [105] |
| Oral | TMC (Cationic polymers) | Modified chitosan (crustaceans, fungi) | Tight junction modulation | In vitro (Caco-2 cells 2) | Mannitol: Sugar alcohol FITC–dextran: Hydrophilic polysaccharide Buserelin: Gonadotropin-releasing hormone agonist | [106] |
| Oral | TMC (Cationic polymers) | Modified chitosan (crustaceans, fungi) | Tight junction modulation | In vitro (Caco-2 cells 2) | Clodronate: Bisphosphonate | [69] |
| Oral | ZOT (Toxins and venom extracts) | Bacteria (Vibrio cholerae) | Tight junction modulation | In vitro (Caco-2 cells 2) | PEG 4000: Polyethylene glycol FITC–dextran: Hydrophilic polysaccharide Inulin: Naturally occurring polysaccharide Paclitaxel: Tetracyclic diterpenoid Acyclovir: HSV-specified DNA polymerases inhibitor Cyclosporine: Immunosuppressant Doxorubicin: Daunorubicin precursor | [107] |
| Pulmonary | Aprotinin, bestatin (Protease inhibitors) | Animal (bovine lung tissue), bacteria (Streptomyces olivoreticuli) | Metabolism inhibition | In vivo (rat) | rhG-CSF: Granulocyte-colony stimulating factor | [108] |
| Pulmonary | Chitosan (Biopolymer) | Chemically modified: deacetylation of chitin Animal (crustaceans), fungi | Tight junction modulation | In vitro (Calu-3 cells 5); in vivo (rat) | Octreotide: Somatostatin analog | [109] |
| Pulmonary | Citric acid (Chelating agents) | Plant (citrus fruits and vegetables), fungi (Aspergillus niger) | Local mucosal tissue modulation; metabolism inhibition | In vivo (rat) | Insulin: Peptide hormone | [110] |
| Pulmonary | HPBCD, Crysmeb (Cyclodextrin derivatives) | Plant (starch) | Tight junction modulation | In vitro (Calu-3 cells 5) | Mannitol: Sugar alcohol | [111] |
| Pulmonary | Lanthanum, cerium, gadolinium (Lanthanides) | Natural elements | Drug targeting | In vivo (rat) | Insulin: Peptide hormone | [112] |
| Pulmonary | Sodium glycocholate (Bile salt) | Intestinal bacterial by-product | Tight junction modulation | Ex vivo (rabbit trachea and jejunum) | Thyrotropin-releasing hormone (TRH): Tripeptidal hypothalamus hormone Insulin: Peptide hormone | [113] |
| Pulmonary | Sodium taurocholate (Bile salt) | Intestinal bacterial by-product | Metabolism enhancement (dissociation of insulin hexamers); tight junction modulation; metabolism (enzymatic degradation) inhibition | In vitro (Caco-2 cells 2), In vivo (dog) | Insulin: Peptide hormone | [114] |
| Pulmonary | Dideoxycytidine: Nucleoside analog reverse transcriptase inhibitor (NRTI) | Plant (Starch) | Tight junction modulation | In vitro (Calu-3 cells 5), in vivo (rat) | Enoxaparin: Anticoagulant | [115] |
| Pulmonary | TMC (Cationic polymers) | Chemically modified: deacetylation of chitin Animal (crustaceans), fungi | Tight junction modulation | In vitro (Calu-3 cells 5); in vivo (rat) | Octreotide: Octapeptide | [109] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterson, B.; Weyers, M.; Steenekamp, J.H.; Steyn, J.D.; Gouws, C.; Hamman, J.H. Drug Bioavailability Enhancing Agents of Natural Origin (Bioenhancers) that Modulate Drug Membrane Permeation and Pre-Systemic Metabolism. Pharmaceutics 2019, 11, 33. https://doi.org/10.3390/pharmaceutics11010033
Peterson B, Weyers M, Steenekamp JH, Steyn JD, Gouws C, Hamman JH. Drug Bioavailability Enhancing Agents of Natural Origin (Bioenhancers) that Modulate Drug Membrane Permeation and Pre-Systemic Metabolism. Pharmaceutics. 2019; 11(1):33. https://doi.org/10.3390/pharmaceutics11010033
Chicago/Turabian StylePeterson, Bianca, Morné Weyers, Jan H. Steenekamp, Johan D. Steyn, Chrisna Gouws, and Josias H. Hamman. 2019. "Drug Bioavailability Enhancing Agents of Natural Origin (Bioenhancers) that Modulate Drug Membrane Permeation and Pre-Systemic Metabolism" Pharmaceutics 11, no. 1: 33. https://doi.org/10.3390/pharmaceutics11010033
APA StylePeterson, B., Weyers, M., Steenekamp, J. H., Steyn, J. D., Gouws, C., & Hamman, J. H. (2019). Drug Bioavailability Enhancing Agents of Natural Origin (Bioenhancers) that Modulate Drug Membrane Permeation and Pre-Systemic Metabolism. Pharmaceutics, 11(1), 33. https://doi.org/10.3390/pharmaceutics11010033