Adjuvant Antitumor Immunity Contributes to the Overall Antitumor Effect of Pegylated Liposomal Doxorubicin (Doxil®) in C26 Tumor-Bearing Immunocompetent Mice

 ,
,  , and
, and
Abstract
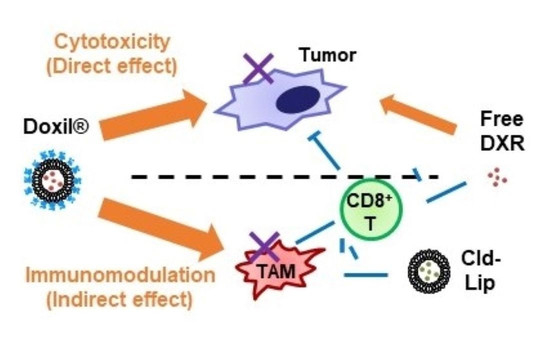
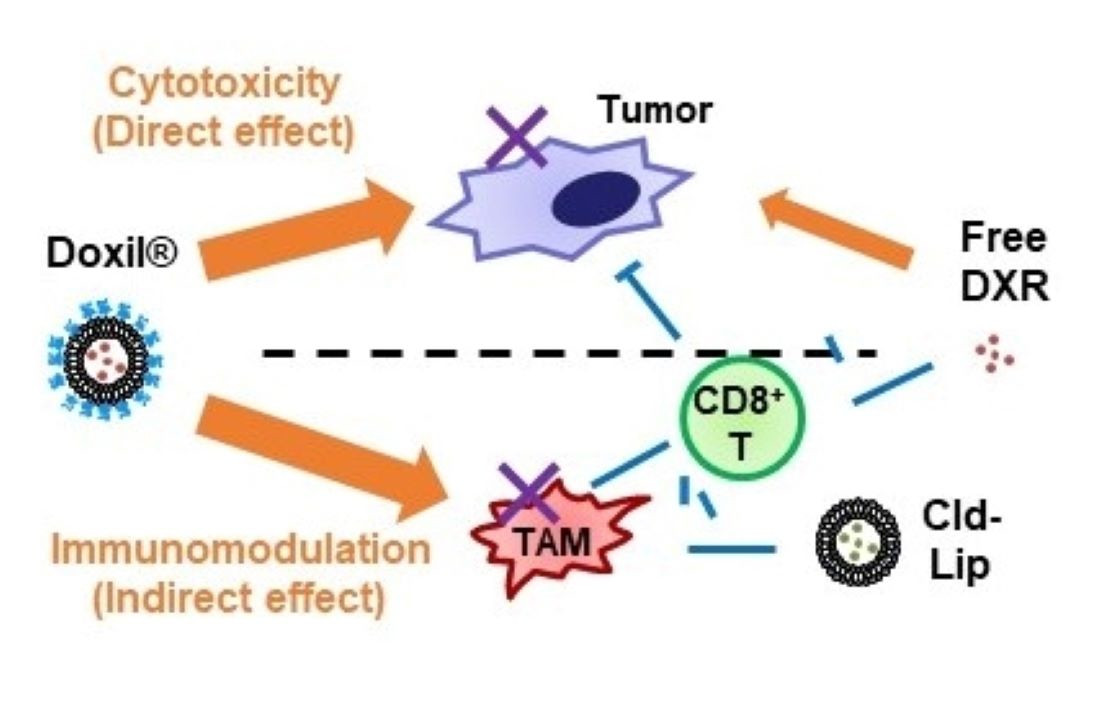
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animal and Tumor Cell Line
2.3. Preparation of Clodronate-Containing Liposomes
2.4. Antitumor Effect of DXR Formulations
2.5. Effect of Treatment with DXR Formulations on Immune Cell Populations in the Tumor Tissue
2.6. Evaluation of MHC Class I Expression on Tumor Cells
2.7. Statistical Analysis
3. Results
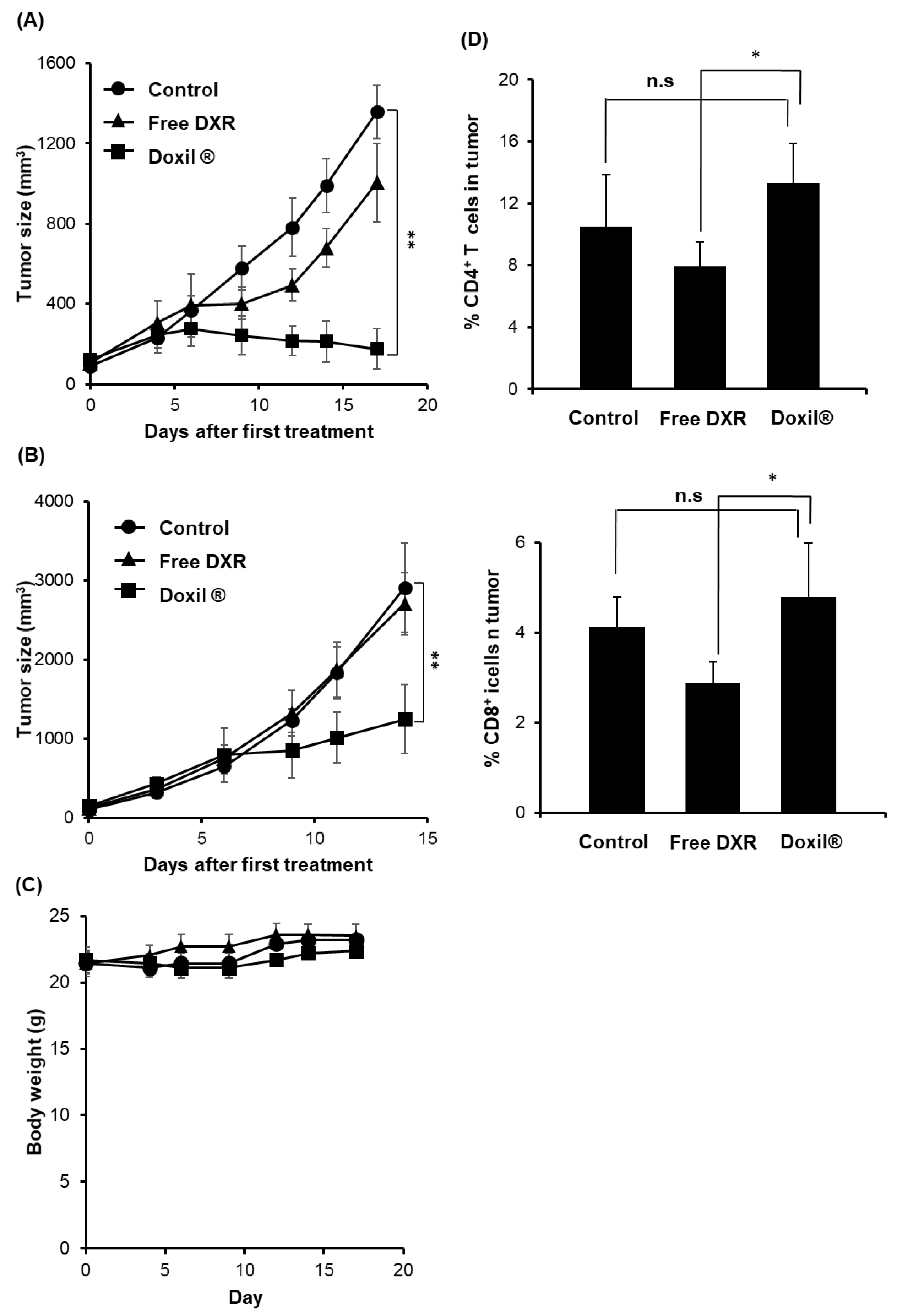
3.1. Tumor Growth Suppressive Effects of DXR Formulations in C26 Tumor-Bearing Immunocompetent Versus Immunodeficient Nude Mouse Models
3.2. Effect of Treatment with Doxil® on Protumor Host Immunity
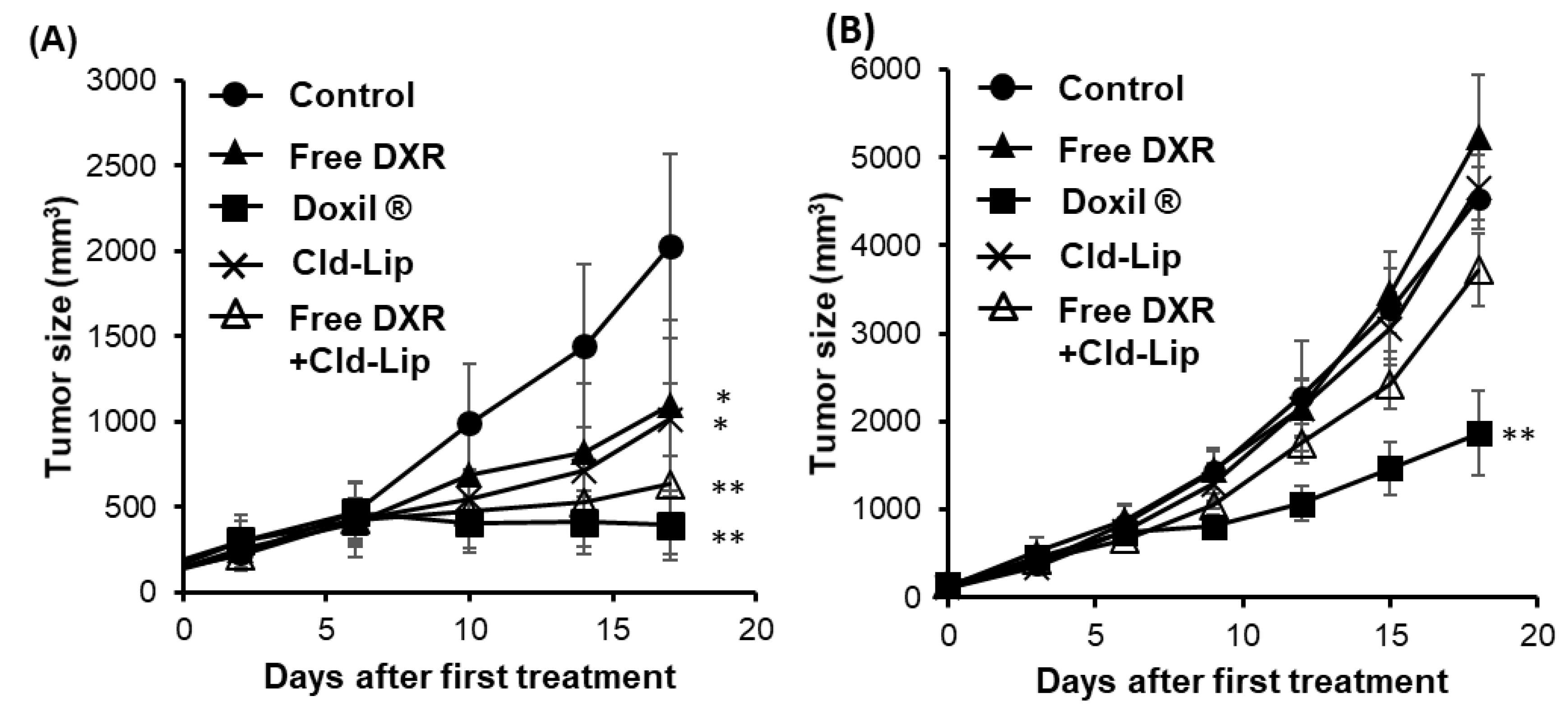
3.3. Effect of Depletion of TAMs in the Tumor Tissues on Doxil®-Mediated Tumor Growth Suppression in Immunocompetent Mice
3.4. Changes Caused by either Doxil® or Free DXR Treatment on MHC I Expression Levels on Tumor Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wehner, R.; Bitterlich, A.; Meyer, N.; Kloss, A.; Schakel, K.; Bachmann, M.; Schmitz, M. Impact of chemotherapeutic agents on the immunostimulatory properties of human 6-sulfo LacNAc+ (slan) dendritic cells. Int. J. Cancer 2013, 132, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Tsuchikawa, T.; Miyamoto, M.; Yamamura, Y.; Shichinohe, T.; Hirano, S.; Kondo, S. The immunological impact of neoadjuvant chemotherapy on the tumor microenvironment of esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2012, 19, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A. Chemoimmunotherapy. Cancer J. 2010, 16, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Combination of chemotherapy and immunotherapy for cancer: A paradigm revisited. Lancet Oncol. 2007, 8, 2–3. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Aoto, K.; Mimura, K.; Okayama, H.; Saito, M.; Chida, S.; Noda, M.; Nakajima, T.; Saito, K.; Abe, N.; Ohki, S.; et al. Immunogenic tumor cell death induced by chemotherapy in patients with breast cancer and esophageal squamous cell carcinoma. Oncol. Rep. 2018, 39, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Lutsiak, M.E.; Semnani, R.T.; De Pascalis, R.; Kashmiri, S.V.; Schlom, J.; Sabzevari, H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005, 105, 2862–2868. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Devaud, C.; John, L.B.; Westwood, J.A.; Darcy, P.K. Enhancing immunotherapy using chemotherapy and radiation to modify the tumor microenvironment. Oncoimmunology 2013, 2, e25962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevko, A.; Michels, T.; Vrohlings, M.; Umansky, L.; Beckhove, P.; Kato, M.; Shurin, G.V.; Shurin, M.R.; Umansky, V. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J. Immunol. 2013, 190, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.S.; Yan, W.L.; Chang, Y.W.; Yeh, Y.C.; Chen, H.W.; Leng, C.H.; Liu, S.J. Gemcitabine enhances antitumor efficacy of recombinant lipoimmunogen-based immunotherapy. Oncoimmunology 2016, 5, e1095433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Abu Lila, A.S.; Nishio, M.; Doi, Y.; Ando, H.; Ukawa, M.; Ishima, Y.; Ishida, T. Modulation of antitumor immunity contributes to the enhanced therapeutic efficacy of liposomal oxaliplatin in mouse model. Cancer Sci. 2017, 108, 1864–1869. [Google Scholar] [CrossRef] [PubMed]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Alizadeh, D.; Trad, M.; Hanke, N.T.; Larmonier, C.B.; Janikashvili, N.; Bonnotte, B.; Katsanis, E.; Larmonier, N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014, 74, 104–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Eralp, Y.; Wang, X.; Wang, J.P.; Maughan, M.F.; Polo, J.M.; Lachman, L.B. Doxorubicin and paclitaxel enhance the antitumor efficacy of vaccines directed against HER 2/neu in a murine mammary carcinoma model. Breast Cancer Res. 2004, 6, R275–R283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panis, C.; Lemos, L.G.; Victorino, V.J.; Herrera, A.C.; Campos, F.C.; Colado Simao, A.N.; Pinge-Filho, P.; Cecchini, A.L.; Cecchini, R. Immunological effects of taxol and adryamicin in breast cancer patients. Cancer Immunol. Immunother. 2012, 61, 481–488. [Google Scholar] [CrossRef]
- Desfrancois, C.; Auzely, R.; Texier, I. Lipid Nanoparticles and Their Hydrogel Composites for Drug Delivery: A Review. Pharmaceuticals 2018, 11, 118. [Google Scholar] [CrossRef] [Green Version]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Diehl, K.H.; Hull, R.; Morton, D.; Pfister, R.; Rabemampianina, Y.; Smith, D.; Vidal, J.M.; van de Vorstenbosch, C. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J. Appl. Toxicol. 2001, 21, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crommelin, D.J.A.; van Hoogevest, P.; Storm, G. The role of liposomes in clinical nanomedicine development. What now? Now what? J. Control. Release 2020, 318, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Moosavian, S.A.; Bianconi, V.; Pirro, M.; Sahebkar, A. Challenges and pitfalls in the development of liposomal delivery systems for cancer therapy. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzyme Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Cabanes, A.; Tzemach, D.; Goren, D.; Horowitz, A.T.; Gabizon, A. Comparative study of the antitumor activity of free doxorubicin and polyethylene glycol-coated liposomal doxorubicin in a mouse lymphoma model. Clin. Cancer Res. 1998, 4, 499–505. [Google Scholar]
- Barenholz, Y. Doxil(R)-the first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Allen, T.M.; Martin, F.J. Advantages of liposomal delivery systems for anthracyclines. Semin. Oncol. 2004, 31, 5–15. [Google Scholar] [CrossRef]
- Storm, G.; van Hoesel, Q.G.; de Groot, G.; Kop, W.; Steerenberg, P.A.; Hillen, F.C. A comparative study on the antitumor effect, cardiotoxicity and nephrotoxicity of doxorubicin given as a bolus, continuous infusion or entrapped in liposomes in the Lou/M Wsl rat. Cancer Chemother. Pharmacol. 1989, 24, 341–348. [Google Scholar] [CrossRef]
- Laginha, K.M.; Verwoert, S.; Charrois, G.J.; Allen, T.M. Determination of doxorubicin levels in whole tumor and tumor nuclei in murine breast cancer tumors. Clin. Cancer Res. 2005, 11, 6944–6949. [Google Scholar] [CrossRef] [Green Version]
- Van Rooijen, N.; Sanders, A. Liposome mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 1994, 174, 83–93. [Google Scholar] [CrossRef]
- El Sayed, M.M.; Takata, H.; Shimizu, T.; Kawaguchi, Y.; Abu Lila, A.S.; Elsadek, N.E.; Alaaeldin, E.; Ishima, Y.; Ando, H.; Kamal, A.; et al. Hepatosplenic phagocytic cells indirectly contribute to anti-PEG IgM production in the accelerated blood clearance (ABC) phenomenon against PEGylated liposomes: Appearance of an unexplained mechanism in the ABC phenomenon. J. Control. Release 2020, 323, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Abu Lila, A.S.; Kizuki, S.; Doi, Y.; Suzuki, T.; Ishida, T.; Kiwada, H. Oxaliplatin encapsulated in PEG-coated cationic liposomes induces significant tumor growth suppression via a dual-targeting approach in a murine solid tumor model. J. Control. Release 2009, 137, 8–14. [Google Scholar] [CrossRef]
- ElBayoumi, T.A.; Torchilin, V.P. Tumor-targeted nanomedicines: Enhanced antitumor efficacy in vivo of doxorubicin-loaded, long-circulating liposomes modified with cancer-specific monoclonal antibody. Clin. Cancer Res. 2009, 15, 1973–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, H.; Sangai, T.; Ishii, G.; Ikehara, A.; Nagashima, T.; Miyazaki, M.; Ochiai, A. Stromal MCP-1 in mammary tumors induces tumor-associated macrophage infiltration and contributes to tumor progression. Int. J. Cancer. 2009, 125, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Harashima, H.; Kiwada, H. Liposome clearance. Biosci. Rep. 2002, 22, 197–224. [Google Scholar] [CrossRef] [PubMed]
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjallman, A.H.; Ballmer-Hofer, K.; Schwendener, R.A. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br. J. Cancer. 2006, 95, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Ruiz-Cabello, F.; Cabrera, T.; Perez-Villar, J.J.; Lopez-Botet, M.; Duggan-Keen, M.; Stern, P.L. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol. Today 1997, 18, 89–95. [Google Scholar] [CrossRef]
- Predina, J.D.; Judy, B.; Aliperti, L.A.; Fridlender, Z.G.; Blouin, A.; Kapoor, V.; Laguna, B.; Nakagawa, H.; Rustgi, A.K.; Aguilar, L.; et al. Neoadjuvant in situ gene-mediated cytotoxic immunotherapy improves postoperative outcomes in novel syngeneic esophageal carcinoma models. Cancer Gene. Ther. 2011, 18, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.D.; Headley, M.B.; Hodzic, A.; Walsh, G.M.; Wingett, D.G. In vitro and in vivo immunosuppressive activity of a novel anthracycline, 13-deoxy, 5-iminodoxorubicin. Int. Immunopharmacol. 2007, 7, 734–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yu, X.; Wang, Z.; Wu, P.; Huang, J. Anthracyclines potentiate anti-tumor immunity: A new opportunity for chemoimmunotherapy. Cancer Lett. 2015, 369, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Schioppa, T.; Porta, C.; Allavena, P.; Sica, A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006, 25, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhi, Q.; Zhou, B.P.; Tao, M.; Liu, J.; Li, W. The Role of Tumor Associated Macrophages in the Tumor Microenvironment: Mechanism and Functions. Anticancer Agents Med. Chem. 2016, 16, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Takeya, M.; Komohara, Y. Role of tumor-associated macrophages in human malignancies: Friend or foe? Pathol. Int. 2016, 66, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y. Tumor-associated macrophages, potential targets for cancer treatment. Biomark. Res. 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef]
- Malesci, A.; Bianchi, P.; Celesti, G.; Basso, G.; Marchesi, F.; Grizzi, F.; Di Caro, G.; Cavalleri, T.; Rimassa, L.; Palmqvist, R.; et al. Tumor-associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology 2017, 6, e1342918. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.M.; Tennis, M.A.; Orlicky, D.J.; Lin, H.; Ju, C.; Redente, E.F.; Choo, K.S.; Staab, T.A.; Bouchard, R.J.; Merrick, D.T.; et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front. Immunol. 2014, 5, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takayama, T.; Shimizu, T.; Abu Lila, A.S.; Kanazawa, Y.; Ando, H.; Ishima, Y.; Ishida, T. Adjuvant Antitumor Immunity Contributes to the Overall Antitumor Effect of Pegylated Liposomal Doxorubicin (Doxil®) in C26 Tumor-Bearing Immunocompetent Mice. Pharmaceutics 2020, 12, 990. https://doi.org/10.3390/pharmaceutics12100990
Takayama T, Shimizu T, Abu Lila AS, Kanazawa Y, Ando H, Ishima Y, Ishida T. Adjuvant Antitumor Immunity Contributes to the Overall Antitumor Effect of Pegylated Liposomal Doxorubicin (Doxil®) in C26 Tumor-Bearing Immunocompetent Mice. Pharmaceutics. 2020; 12(10):990. https://doi.org/10.3390/pharmaceutics12100990
Chicago/Turabian StyleTakayama, Takuma, Taro Shimizu, Amr S. Abu Lila, Yuki Kanazawa, Hidenori Ando, Yu Ishima, and Tatsuhiro Ishida. 2020. "Adjuvant Antitumor Immunity Contributes to the Overall Antitumor Effect of Pegylated Liposomal Doxorubicin (Doxil®) in C26 Tumor-Bearing Immunocompetent Mice" Pharmaceutics 12, no. 10: 990. https://doi.org/10.3390/pharmaceutics12100990
APA StyleTakayama, T., Shimizu, T., Abu Lila, A. S., Kanazawa, Y., Ando, H., Ishima, Y., & Ishida, T. (2020). Adjuvant Antitumor Immunity Contributes to the Overall Antitumor Effect of Pegylated Liposomal Doxorubicin (Doxil®) in C26 Tumor-Bearing Immunocompetent Mice. Pharmaceutics, 12(10), 990. https://doi.org/10.3390/pharmaceutics12100990