Pulmonary Delivery of Biological Drugs




Abstract
:
1. Introduction
2. Structure and Characteristics of Biological Drugs
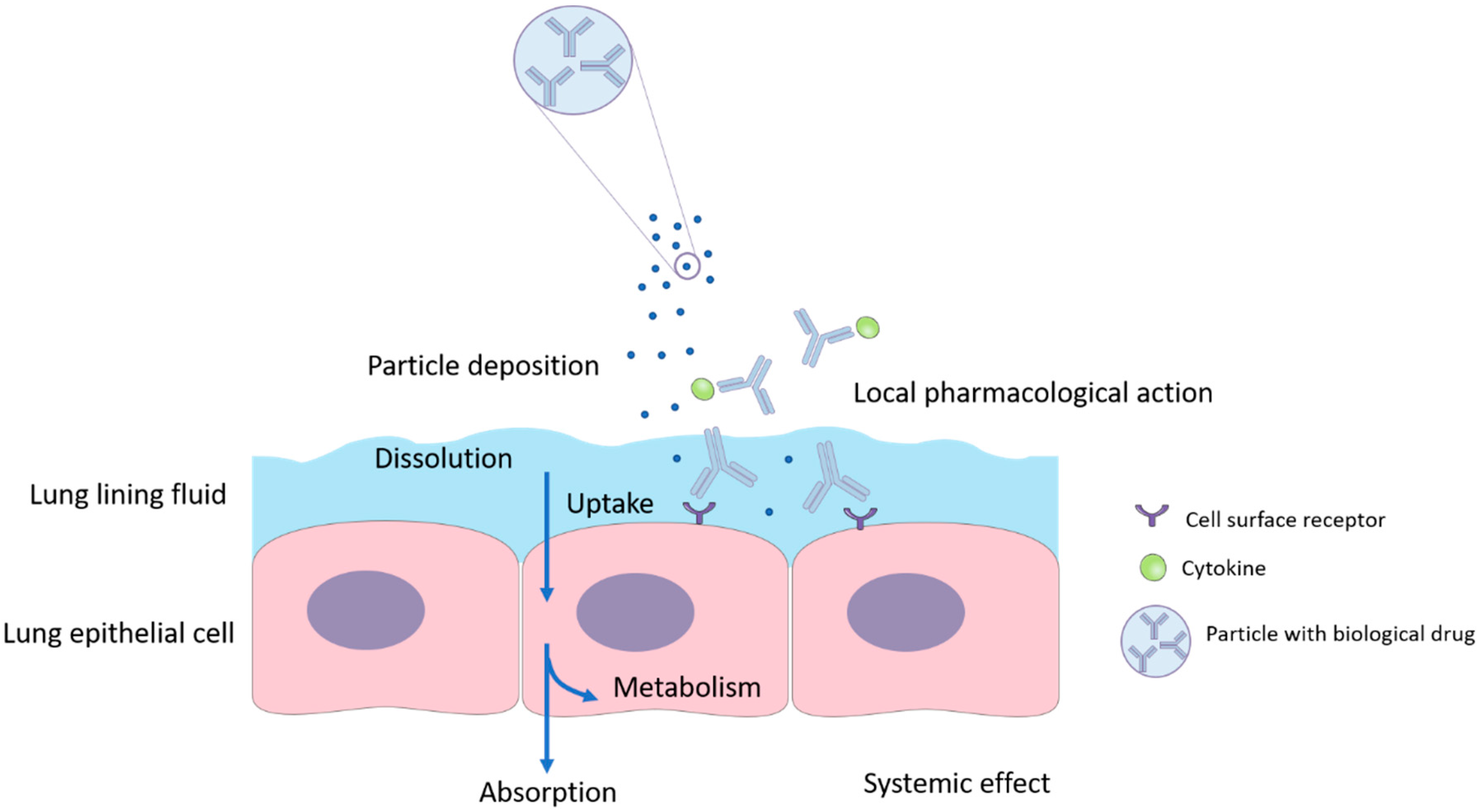
3. Challenges in Pulmonary Delivery of Biological Drugs
3.1. Anatomical Barriers
3.2. Mucociliary and Macrophage Clearance
3.3. Pulmonary Surfactant
3.4. Airway Epithelium
3.5. Metabolism
4. Strategies for Inhaled Delivery of Biological Drugs
4.1. Antibody Fragments
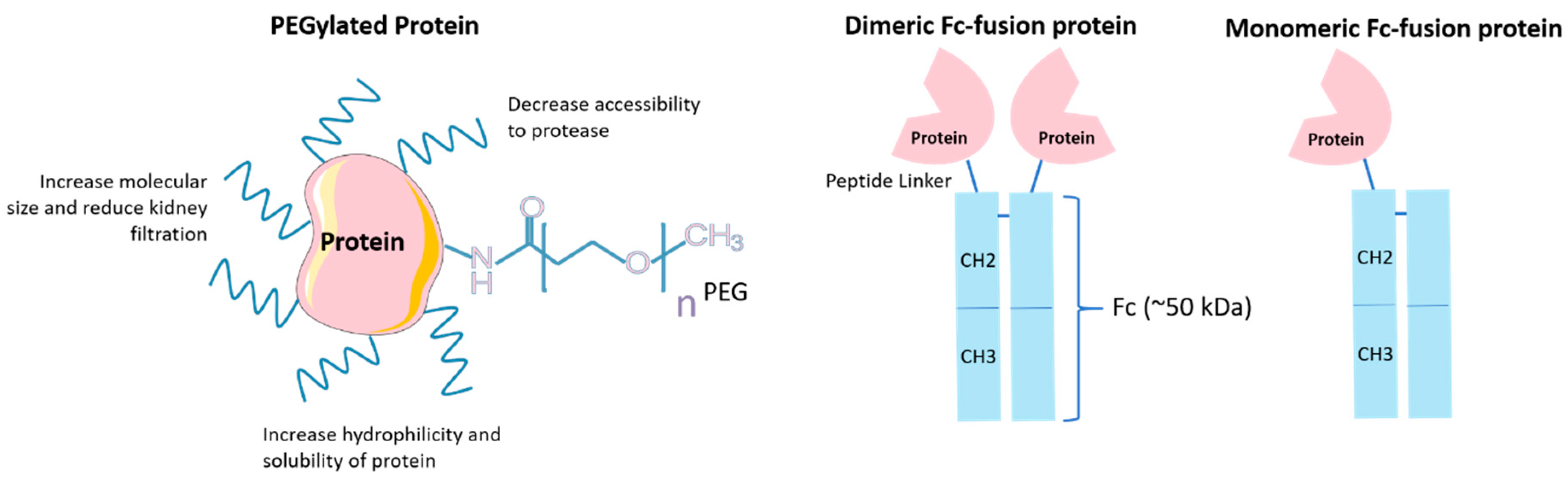
4.2. PEGylation
4.3. Fc Engineering
5. Inhalation Technology
5.1. Nebulisation
5.2. Pressurised Metred-Dose Inhalers (pMDIs)
5.3. Dry Powder Inhalers (DPIs)
6. Clinical Developments
6.1. Inhaled Therapeutic Proteins and Peptides
6.2. Inhaled mAbs and Antibody Fragments
7. Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Zelikin, A.N.; Healy, C.E.A.M. Materials and methods for delivery of biological drugs. Nat. Chem. 2016, 8, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Durán-Lobato, M.; Niu, Z.; Alonso, M.J.J.A.M. Oral delivery of biologics for precision medicine. Adv. Mater. 2020, 32, 1901935. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2019. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2020, 25, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, L.J.; Kim, E.S. Emicizumab-kxwh: First global approval. Drugs 2018, 78, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.B.; Lebwohl, M.G. Review of safety and efficacy of approved systemic psoriasis therapies. Int. J. Dermatol. 2019, 58, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Suzman, D.L.; Blumenthal, G.; Mushti, S.; He, K.; Libeg, M.; Keegan, P.; Pazdur, R. FDA Approval Summary: Nivolumab for the Treatment of Metastatic Non-Small Cell Lung Cancer With Progression On or After Platinum-Based Chemotherapy. Oncologist 2016, 21, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Fala, L. Nucala (mepolizumab): First IL-5 antagonist monoclonal antibody FDA approved for maintenance treatment of patients with severe asthma. Am. Health Drug Benefits 2016, 9, 106–110. [Google Scholar]
- Saco, T.V.; Pepper, A.N.; Lockey, R.F. Benralizumab for the treatment of asthma. Expert Rev. Clin. Immunol. 2017, 13, 405–413. [Google Scholar] [CrossRef]
- Shirley, M. Dupilumab: First global approval. Drugs 2017, 77, 1115–1121. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Gokarn, Y.; Mitragotri, S. Non-invasive delivery strategies for biologics. Nat. Rev. Drug Discov. 2019, 18, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Hil-Lal, T.A.; Byun, Y. Strategies for non-invasive delivery of biologics. J. Drug Target. 2012, 20, 481–501. [Google Scholar] [CrossRef] [PubMed]
- Griese, M.; Scheuch, G. Delivery of Alpha-1 Antitrypsin to Airways. Ann. Am. Thorac. Soc. 2016, 13, S346–S351. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, C.J.; Giovannitti, J.A.; Boynes, S.G. Needle Phobia: Etiology, Adverse Consequences, and Patient Management. Dent. Clin. 2010, 54, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Vllasaliu, D.; Thanou, M.; Stolnik, S.; Fowler, R. Recent advances in oral delivery of biologics: Nanomedicine and physical modes of delivery. Expert Opin. Drug Deliv. 2018, 15, 759–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montenegro-Nicolini, M.; Morales, J.O. Overview and future potential of buccal mucoadhesive films as drug delivery systems for biologics. AAPS PharmSciTech 2017, 18, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, J.; Lupo, N.; Bernkop-Schnürch, A. Advanced formulations for intranasal delivery of biologics. Int. J. Pharm. 2018, 553, 8–20. [Google Scholar] [CrossRef]
- Ferrati, S.; Wu, T.; Kanapuram, S.R.; Smyth, H.D. Dosing considerations for inhaled biologics. Int. J. Pharm. 2018, 549, 58–66. [Google Scholar] [CrossRef]
- Morales, J.O.; Fathe, K.R.; Brunaugh, A.; Ferrati, S.; Li, S.; Montenegro-Nicolini, M.; Mousavikhamene, Z.; McConville, J.T.; Prausnitz, M.R.; Smyth, H.D.C. Challenges and Future Prospects for the Delivery of Biologics: Oral Mucosal, Pulmonary, and Transdermal Routes. AAPS J. 2017, 19, 652–668. [Google Scholar] [CrossRef]
- Agu, R.; Ugwoke, M.I.; Armand, M.; Kinget, R.; Verbeke, N. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir. Res. 2001, 2, 198–209. [Google Scholar]
- Guilleminault, L.; Azzopardi, N.; Arnoult, C.; Sobilo, J.; Hervé, V.; Montharu, J.; Guillon, A.; Andres, C.; Hérault, O.; Le Pape, A.; et al. Fate of inhaled monoclonal antibodies after the deposition of aerosolized particles in the respiratory system. J. Control. Release 2014, 196, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Maillet, A.; Guilleminault, L.; Lemarie, E.; Lerondel, S.; Azzopardi, N.; Montharu, J.; Congy-Jolivet, N.; Reverdiau, P.; Legrain, B.; Parent, C.; et al. The Airways, a Novel Route for Delivering Monoclonal Antibodies to Treat Lung Tumors. Pharm. Res. 2011, 28, 2147–2156. [Google Scholar] [CrossRef]
- Gänsslen, M. Über inhalation von insulin. J. Mol. Med. 1925, 4, 71. [Google Scholar] [CrossRef]
- Patton, J.S.; Byron, P.R. Inhaling medicines: Delivering drugs to the body through the lungs. Nature Rev. Drug Discov. 2007, 6, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.S.; McCabe, J.G.; Hansen, S.E.; Daugherty, A.L. Absorption of human growth hormone from the rat lung. Biotechnol. Ther. 1989, 1, 213–228. [Google Scholar] [PubMed]
- Labiris, N.; Dolovich, M. Pulmonary drug delivery. Part I: Physiological factors affecting therapeutic effectiveness of aerosolized medications. Br. J. Clin. Pharmacol. 2003, 56, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Van Heeke, G.; Allosery, K.; De Brabandere, V.; De Smedt, T.; Detalle, L.; Fougerolles, A. Nanobodies® as inhaled biotherapeutics for lung diseases. Pharm. Ther. 2017, 169, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Depreter, F.; Pilcer, G.; Amighi, K. Inhaled proteins: Challenges and perspectives. Int. J. Pharm. 2013, 447, 251–280. [Google Scholar] [CrossRef]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Buss, N.A.; Henderson, S.J.; McFarlane, M.; Shenton, J.M.; De Haan, L. Monoclonal antibody therapeutics: History and future. Curr. Opin. Pharmacol. 2012, 12, 615–622. [Google Scholar] [CrossRef]
- Booth, B.J.; Ramakrishnan, B.; Narayan, K.; Wollacott, A.M.; Babcock, G.J.; Shriver, Z.; Viswanathan, K. Extending human IgG half-life using structure-guided design. mAbs 2018, 10, 1098–1110. [Google Scholar] [CrossRef] [PubMed]
- Grevys, A.; Nilsen, J.; Sand, K.M.K.; Daba, M.B.; Øynebråten, I.; Bern, M.; McAdam, M.B.; Foss, S.; Schlothauer, T.; Michaelsen, T.E.; et al. A human endothelial cell-based recycling assay for screening of FcRn targeted molecules. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillet, A.; Congy-Jolivet, N.; Le Guellec, S.; Vecellio, L.; Hamard, S.; Courty, Y.; Courtois, A.; Gauthier, F.; Diot, P.; Thibault, G.; et al. Aerodynamical, Immunological and Pharmacological Properties of the Anticancer Antibody Cetuximab Following Nebulization. Pharm. Res. 2008, 25, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, H.; Najafabadi, A.R.; Daman, Z.; Ghasemian, E.; Montazeri, H.; Vatanara, A. Respiratory Administration of Infliximab Dry Powder for Local Suppression of Inflammation. AAPS PharmSciTech 2019, 20, 128. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V.; Cockcroft, D.W.; Boulet, L.-P.; Wong, H.H.; Deschesnes, F.; Davis, E.E.; Ruppel, J.; Su, J.Q.; Adelman, D.C. Effect of Aerosolized Anti-IgE (E25) on Airway Responses to Inhaled Allergen in Asthmatic Subjects. Am. J. Respir. Crit. Care Med. 1999, 160, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Respaud, R.; Marchand, D.; Pelat, T.; Tchou-Wong, K.-M.; Roy, C.J.; Parent, C.; Cabrera, M.; Guillemain, J.; MacLoughlin, R.; Levacher, E.; et al. Development of a drug delivery system for efficient alveolar delivery of a neutralizing monoclonal antibody to treat pulmonary intoxication to ricin. J. Control. Release 2016, 234, 21–32. [Google Scholar] [CrossRef]
- Freches, D.; Patil, H.P.; Franco, M.M.; Uyttenhove, C.; Heywood, S.; Vanbever, R. PEGylation prolongs the pulmonary retention of an anti-IL-17A Fab’ antibody fragment after pulmonary delivery in three different species. Int. J. Pharm. 2017, 521, 120–129. [Google Scholar] [CrossRef]
- Proudfoot, A.; Bayliffe, A.; O’Kane, C.M.; Wright, T.; Serone, A.; Bareille, P.J.; Brown, V.; Hamid, U.I.; Chen, Y.; Wilson, R.; et al. Novel anti-tumour necrosis factor receptor-1 (TNFR1) domain antibody prevents pulmonary inflammation in experimental acute lung injury. Thorax 2018, 73, 723–730. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Nema, S.; Teagarden, D. Protein aggregation-pathways and influencing factors. Int. J. Pharm. 2010, 390, 89–99. [Google Scholar] [CrossRef]
- Wang, W.; Roberts, C.J. Protein aggregation—Mechanisms, detection, and control. Int. J. Pharm. 2018, 550, 251–268. [Google Scholar] [CrossRef]
- Castillo, G.M.; Ngo, C.; Cummings, J.; Wight, T.N.; Snow, A.D. Perlecan binds to the β-amyloid proteins (Aβ) of Alzheimer’s disease, accelerates Aβ fibril formation, and maintains Aβ fibril stability. J. Neurochem. 1997, 69, 2452–2465. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Clos, A.L.; Midoro-Hiriuti, T.; Goldblum, R.M.; Jackson, G.R.; Kayed, R. Inhaled Insulin Forms Toxic Pulmonary Amyloid Aggregates. Endocrinology 2010, 151, 4717–4724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strand, V.; Balsa, A.; Al-Saleh, J.; Barile-Fabris, L.; Horiuchi, T.; Takeuchi, T.; Lula, S.; Hawes, C.; Kola, B.; Marshall, L. Immunogenicity of Biologics in Chronic Inflammatory Diseases: A Systematic Review. BioDrugs 2017, 31, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Dingman, R.; Balu-Iyer, S.V. Immunogenicity of Protein Pharmaceuticals. J. Pharm. Sci. 2019, 108, 1637–1654. [Google Scholar] [CrossRef]
- Burgess, G.; Boyce, M.; Jones, M.; Larsson, L.; Main, M.J.; Morgan, F.; Phillips, P.; Scrimgeour, A.; Strimenopoulou, F.; Vajjah, P.; et al. Randomized study of the safety and pharmacodynamics of inhaled interleukin-13 monoclonal antibody fragment VR942. EBioMedicine 2018, 35, 67–75. [Google Scholar] [CrossRef]
- Joo, W.D.; Visintin, I.; Mor, G. Targeted cancer therapy—Are the days of systemic chemotherapy numbered? Maturitas 2013, 76, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetteh, E.; Morris, S. Systematic review of drug administration costs and implications for biopharmaceutical manufacturing. Appl. Health Econ. Health Policy 2013, 11, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Tetteh, E.K.; Morris, S.J.H.E.R. Evaluating the administration costs of biologic drugs: Development of a cost algorithm. Appl. Health Econ. Health Policy 2013, 11, 445–456. [Google Scholar] [CrossRef]
- Pilcer, G.; Amighi, K. Formulation strategy and use of excipients in pulmonary drug delivery. Int. J. Pharm. 2010, 392, 1–19. [Google Scholar] [CrossRef]
- Carvalho, T.C.; Peters, J.I.; Williams, R.O., III. Influence of particle size on regional lung deposition—What evidence is there? Int. J. Pharm. 2011, 406, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.-W.; Liang, W.; Chan, H.-K. Pulmonary delivery of therapeutic siRNA. Adv. Drug Deliv. Rev. 2012, 64, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.S.; Brain, J.D.; Davies, L.A.; Fiegel, J.; Gumbleton, M.; Kim, K.-J.; Sakagami, M.; Vanbever, R.; Ehrhardt, C. The Particle has Landed—Characterizing the Fate of Inhaled Pharmaceuticals. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, S71–S87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruge, C.A.; Kirch, J.; Lehr, C.-M. Pulmonary drug delivery: From generating aerosols to overcoming biological barriers—Therapeutic possibilities and technological challenges. Lancet Respir. Med. 2013, 1, 402–413. [Google Scholar] [CrossRef]
- Groneberg, D.; Witt, C.; Wagner, U.; Chung, K.; Fischer, A. Fundamentals of pulmonary drug delivery. Respir. Med. 2003, 97, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codrons, V.; Vanderbist, F.; Ucakar, B.; Préat, V.; Vanbever, R. Impact of formulation and methods of pulmonary delivery on absorption of parathyroid hormone (1–34) from rat lungs. J. Pharm. Sci. 2004, 93, 1241–1252. [Google Scholar] [CrossRef]
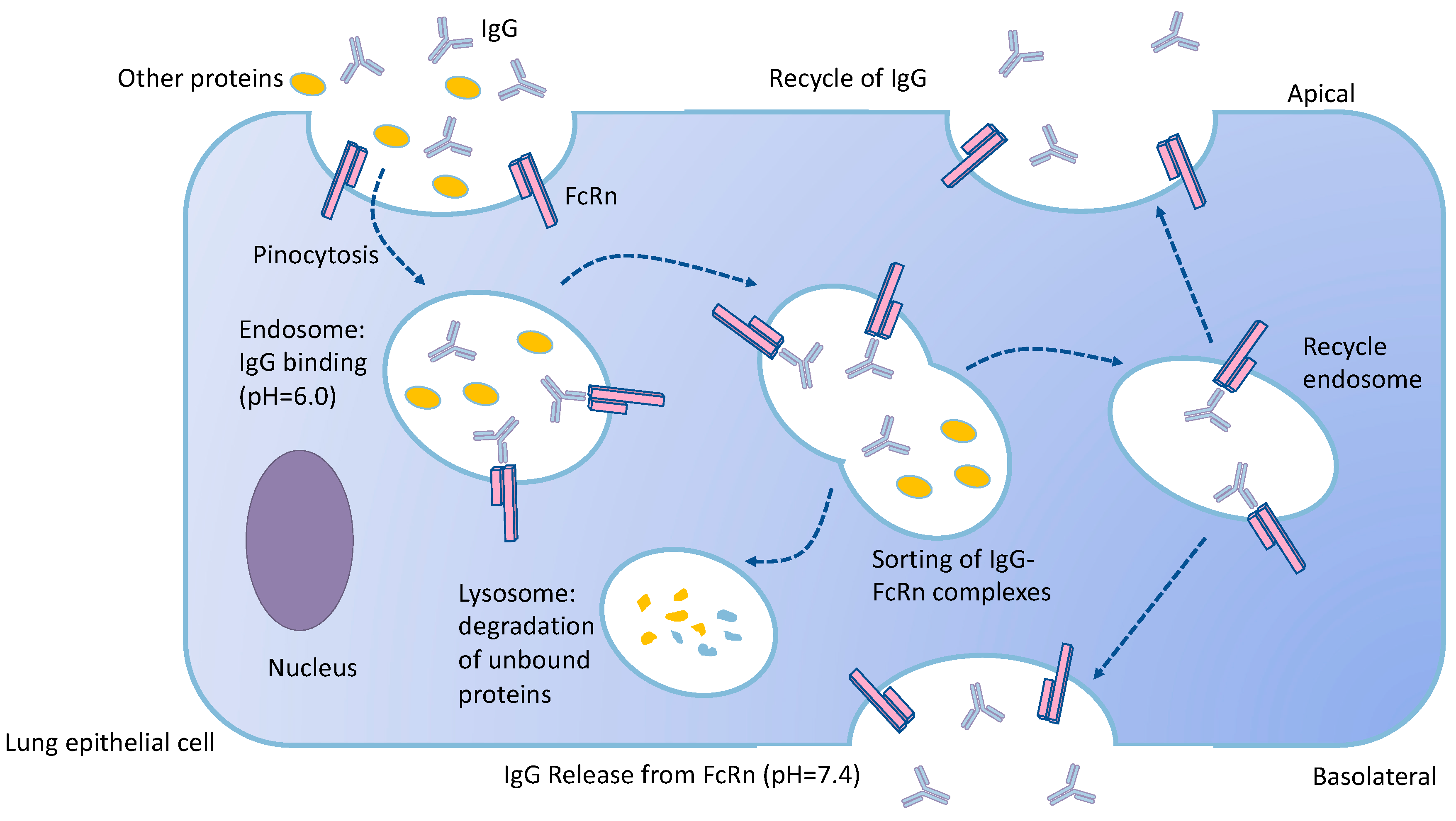
- Spiekermann, G.M.; Finn, P.W.; Ward, E.S.; Dumont, J.; Dickinson, B.L.; Blumberg, R.S.; Lencer, W.I. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: Functional expression of FcRn in the mammalian lung. J. Exp. Med. 2002, 196, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Bitonti, A.J.; Dumont, J.A.; Low, S.C.; Peters, R.T.; Kropp, K.E.; Palombella, V.J.; Stattel, J.M.; Lu, Y.; Tan, C.A.; Song, J.J.; et al. Pulmonary delivery of an erythropoietin Fc fusion protein in non-human primates through an immunoglobulin transport pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 9763–9768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, B.; Bondesson, E.; Borgström, L.; Edsbäcker, E.; Eirefelt, S.; Ekelund, K.; Gustavsson, L.; Hegelund-Myrbäck, T. Pulmonary drug metabolism, clearance and absorption. In Controlled Pulmonary Drug Delivery; Springer: New York, NY, USA, 2011; pp. 21–50. [Google Scholar]
- Munkholm, M.; Mortensen, J. Mucociliary clearance: Pathophysiological aspects. Clin. Physiol. Funct. Imaging 2014, 34, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Patil, H.P.; Freches, D.; Karmani, L.; Duncan, G.A.; Ucakar, B.; Suk, J.S.; Hanes, J.; Gallez, B.; Vanbever, R. Fate of PEGylated antibody fragments following delivery to the lungs: Influence of delivery site, PEG size and lung inflammation. J. Control. Release 2018, 272, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Bøgh, M.; Foged, C.; Müllertz, A.; Nielsen, H.M. Mucosal drug delivery: Barriers, in vitro models and formulation strategies. J. Drug Deliv. Sci. Technol. 2013, 23, 383–391. [Google Scholar] [CrossRef]
- Cone, R.A. Barrier properties of mucus. Adv. Drug Deliv. Rev. 2009, 61, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Olmsted, S.S.; Padgett, J.L.; Yudin, A.I.; Whaley, K.J.; Moench, T.R.; Cone, R.A. Diffusion of Macromolecules and Virus-Like Particles in Human Cervical Mucus. Biophys. J. 2001, 81, 1930–1937. [Google Scholar] [CrossRef] [Green Version]
- Ducreux, J.; Vanbever, R. Crucial biopharmaceutical issues facing macromolecular candidates for inhalation: The role of macrophages in pulmonary protein clearance. Respir. Drug Deliv. Eur. 2007, 2007, 31–41. [Google Scholar]
- Bosquillon, C.; Préat, V.; Vanbever, R. Pulmonary delivery of growth hormone using dry powders and visualization of its local fate in rats. J. Control. Release 2004, 96, 233–244. [Google Scholar] [CrossRef]
- Lombry, C.; Edwards, D.A.; Préat, V.; Vanbever, R. Alveolar macrophages are a primary barrier to pulmonary absorption of macromolecules. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L1002–L1008. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, A.; Cruz, A.; Perez-Gil, J. Barrier or carrier? Pulmonary surfactant and drug delivery. Eur. J. Pharm. Biopharm. 2015, 95, 117–127. [Google Scholar] [CrossRef]
- Guagliardo, R.; Perez-Gil, J.; De Smedt, S.C.; Raemdonck, K. Pulmonary surfactant and drug delivery: Focusing on the role of surfactant proteins. J. Control. Release 2018, 291, 116–126. [Google Scholar] [CrossRef]
- Patton, J.S.; Fishburn, C.S.; Weers, J.G. The Lungs as a Portal of Entry for Systemic Drug Delivery. Proc. Am. Thorac. Soc. 2004, 1, 338–344. [Google Scholar] [CrossRef]
- Ruge, C.A.; Schaefer, U.F.; Herrmann, J.; Kirch, J.; Canadas, O.; Echaide, M. The interplay of lung surfactant proteins and lipids assimilates the macrophage clearance of nanoparticles. PLoS ONE 2012, 7, e40775. [Google Scholar] [CrossRef]
- Zheng, J.; Zheng, Y.; Chen, J.; Fang, F.; He, J.; Li, N.; Tang, Y.; Zhu, J.; Chen, X. Enhanced pulmonary absorption of recombinant human insulin by pulmonary surfactant and phospholipid hexadecanol tyloxapol through Calu-3 monolayers. Pharmazie 2012, 67, 448–451. [Google Scholar]
- Mitra, R.; Pezron, I.; Li, Y.; Mitra, A.K. Enhanced pulmonary delivery of insulin by lung lavage fluid and phospholipids. Int. J. Pharm. 2001, 217, 25–31. [Google Scholar] [CrossRef]
- Kim, K.-J.; Malik, A.B. Protein transport across the lung epithelial barrier. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L247–L259. [Google Scholar] [CrossRef] [Green Version]
- Patton, J.S. Mechanisms of macromolecule absorption by the lungs. Adv. Drug Deliv. Rev. 1996, 19, 3–36. [Google Scholar] [CrossRef]
- Todoroff, J.; Vanbever, R. Fate of nanomedicines in the lungs. Curr. Opin. Colloid Interface Sci. 2011, 16, 246–254. [Google Scholar] [CrossRef]
- John, T.A.; Vogel, S.M.; Minshall, R.D.; Ridge, K.; Tiruppathi, C.; Malik, A.B. Evidence for the role of alveolar epithelial gp60 in active transalveolar albumin transport in the rat lung. J. Physiol. 2001, 533, 547–559. [Google Scholar] [CrossRef]
- Lockett, A.D.; Brown, M.B.; Santos-Falcon, N.; Rush, N.I.; Oueini, H.; Oberle, A.J.; Bolanis, E.; Fragoso, M.A.; Petrusca, D.N.; Serban, K.A.; et al. Active Trafficking of Alpha 1 Antitrypsin across the Lung Endothelium. PLoS ONE 2014, 9, e93979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widera, A.; Kim, K.J.; Crandall, E.D.; Shen, W.C. Transcytosis of GCSF-transferrin across rat alveolar epithelial cell monolayers. Pharm. Res. 2003, 20, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Kawami, M.; Aoki, A.; Yumoto, R. Receptor-mediated endocytosis of macromolecules and strategy to enhance their transport in alveolar epithelial cells. Expert Opin. Drug Deliv. 2015, 12, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Rath, T.; Baker, K.; Dumont, J.A.; Peters, R.T.; Jiang, H.; Qiao, S.-W.; Lencer, W.I.; Pierce, G.F.; Blumberg, R.S. Fc-fusion proteins and FcRn: Structural insights for longer-lasting and more effective therapeutics. Crit. Rev. Biotechnol. 2015, 35, 235–254. [Google Scholar] [CrossRef]
- Low, S.; Nunes, S.; Bitonti, A.; Dumont, J. Oral and pulmonary delivery of FSH–Fc fusion proteins via neonatal Fc receptor-mediated transcytosis. Hum. Reprod. 2005, 20, 1805–1813. [Google Scholar] [CrossRef] [Green Version]
- Vallee, S.; Rakhe, S.; Reidy, T.; Walker, S.; Lu, Q.; Sakorafas, P.; Low, S.; Bitonti, A. Pulmonary Administration of Interferon Beta-1a-Fc Fusion Protein in Non-Human Primates Using an Immunoglobulin Transport Pathway. J. Interf. Cytokine Res. 2012, 32, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Angelo, R.; Rousseau, K.; Grant, M.; Leone-Bay, A.; Richardson, P. Technosphere® Insulin: Defining the Role of Technosphere Particles at the Cellular Level. J. Diabetes Sci. Technol. 2009, 3, 545–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikehata, M.; Yumoto, R.; Nakamura, K.; Nagai, J.; Takano, M. Comparison of Albumin Uptake in Rat Alveolar Type II and Type I-like Epithelial Cells in Primary Culture. Pharm. Res. 2008, 25, 913–922. [Google Scholar] [CrossRef]
- Ma, H.; O’Kennedy, R. Recombinant antibody fragment production. Methods 2017, 116, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.A.; Landon, J. A protocol for ‘enhanced pepsin digestion’: A step by step method for obtaining pure antibody fragments in high yield from serum. J. Immunol. Methods 2003, 275, 239–250. [Google Scholar] [CrossRef]
- Nelson, A.L. Antibody fragments: Hope and hype. mAbs 2010, 2, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacha, J.; Tomlinson, K.; Maertens, L.; Paulissen, G.; Rocks, N.; Foidart, J.-M.; Noël, A.; Palframan, R.; Guéders, M.; Cataldo, D.D. Nebulized Anti–IL-13 Monoclonal Antibody Fab′ Fragment Reduces Allergen-Induced Asthma. Am. J. Respir. Cell Mol. Biol. 2012, 47, 709–717. [Google Scholar] [CrossRef]
- Gozzard, N.; Lightwood, D.; Tservistas, M.; Zehentleitner, M.; Sarkar, K.; Turner, A.; Smith, B.; Lamour, S.D.; Bourne, T.; Shaw, S.; et al. Novel inhaled delivery of anti-IL-13 MAb (FAb fragment): Preclinical efficacy in allergic asthma. In Proceedings of the European Respiratory Society Annual Congress, Milan, Italy, 9–13 September 2017. [Google Scholar]
- Holt, L.J.; Herring, C.; Jespers, L.S.; Woolven, B.P.; Tomlinson, I.M. Domain antibodies: Proteins for therapy. Trends Biotechnol. 2003, 21, 484–490. [Google Scholar] [CrossRef]
- Bertok, S.; Wilson, M.R.; Morley, P.J.; De Wildt, R.; Bayliffe, A.; Takata, M. Selective inhibition of intra-alveolar p55 TNF receptor attenuates ventilator-induced lung injury. Thorax 2012, 67, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordy, J.; Morley, P.J.; Wright, T.J.; Birchler, M.A.; Lewis, A.P.; Emmins, R.; Chen, Y.Z.; Powley, W.M.; Bareille, P.J.; Wilson, R.; et al. Specificity of human anti-variable heavy (VH) chain autoantibodies and impact on the design and clinical testing of a VH domain antibody antagonist of tumour necrosis factor-α receptor 1. Clin. Exp. Immunol. 2015, 182, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.; Bayliffe, A.I.; McAuley, D.F.; Yeung, J.; Thickett, D.R.; Howells, P.A.; O’Donnell, C.; Vassallo, A.M.; Wright, T.J.; McKie, E.; et al. A randomised, placebo-controlled pilot study of a nebulised antitumour necrosis factor receptor-1 domain antibody in patients at risk of postoperative lung injury. Eur. J. Anaesthesiol. 2020, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Ann. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef] [PubMed]
- Detalle, L.; Stohr, T.; Palomo, C.; Piedra, P.A.; Gilbert, B.E.; Mas, V.; Millar, A.; Power, U.F.; Stortelers, C.; Allosery, K.; et al. Generation and Characterization of ALX-0171, a Potent Novel Therapeutic Nanobody for the Treatment of Respiratory Syncytial Virus Infection. Antimicrob. Agents Chemother. 2016, 60, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.L.; Detalle, L.; Gallup, J.M.; Van Geelen, A.; Stohr, T.; Duprez, L.; Ackermann, M.R. Delivery of ALX-0171 by inhalation greatly reduces respiratory syncytial virus disease in newborn lambs. mAbs 2018, 10, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Ibañez, L.I.; De Filette, M.; Hultberg, A.; Verrips, T.; Temperton, N.J.; Weiss, R.A.; Vandevelde, W.; Schepens, B.; Vanlandschoot, P.; Saelens, X. Nanobodies With In Vitro Neutralizing Activity Protect Mice Against H5N1 Influenza Virus Infection. J. Infect. Dis. 2011, 203, 1063–1072. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.F.; Bagci, U.; Keith, L.; Tang, X.; Mollura, D.J.; Zeitlin, L.; Qin, J.; Huzella, L.; Bartos, C.J.; Bohorova, N.; et al. 3B11-N, a monoclonal antibody against MERS-CoV, reduces lung pathology in rhesus monkeys following intratracheal inoculation of MERS-CoV Jordan-n3/2012. Virology 2016, 490, 49–58. [Google Scholar] [CrossRef]
- He, L.; Tai, W.; Li, J.; Chen, Y.; Gao, Y.; Li, J.; Sun, S.; Zhou, Y.; Du, L.; Zhao, G. Enhanced Ability of Oligomeric Nanobodies Targeting MERS Coronavirus Receptor-Binding Domain. Viruses 2019, 11, 166. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; He, L.; Sun, S.; Qiu, H.; Tai, W.; Chen, J.; Li, J.; Chen, Y.; Guo, Y.; Wang, Y.; et al. A Novel Nanobody Targeting Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Receptor-Binding Domain Has Potent Cross-Neutralizing Activity and Protective Efficacy against MERS-CoV. J. Virol. 2018, 92, e00837-18. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.I.; Chang, S.; Shim, J.M.; Jin, J.; Lim, C.-S.; Baek, S.; Min, J.-Y.; Park, W.B.; Oh, M.-D.; Chung, J.; et al. Generation of a Nebulizable CDR-Modified MERS-CoV Neutralizing Human Antibody. Int. J. Mol. Sci. 2019, 20, 5073. [Google Scholar] [CrossRef] [Green Version]
- Gai, J.; Ma, L.; Li, G.; Zhu, M.; Qiao, P.; Li, X.; Zhang, H.; Zhang, Y.; Chen, Y.; Ji, W.; et al. A potent neutralizing nanobody against SARS-CoV-2 with inhaled delivery potential. BioRxiv 2020. [Google Scholar] [CrossRef]
- Martinez-Delgado, G. Inhaled nanobodies against COVID-19. Nat. Rev. Immunol. 2020, 20, 593. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, L.; Parke, H.G.; Ferguson, L.J.; Miller, A.; Shields, M.D.; Detalle, L.; Power, U.F. Comparative Therapeutic Potential of ALX-0171 and Palivizumab against Respiratory Syncytial Virus Clinical Isolate Infection of Well-Differentiated Primary Pediatric Bronchial Epithelial Cell Cultures. Antimicrob. Agents Chemother. 2019, 64. [Google Scholar] [CrossRef] [PubMed]
- Detalle, L.; Ackermann, M.R.; Larios, A.; Gallup, J.; Van Geelen, A.; Duprez, L.; Stohr, T. Delivery of ALX-0171 by inhalation greatly reduces disease burden in a neonatal lamb RSV infection model. In Proceedings of the 9th International Respiratory Syncytial Virus Symposium, Cape Town, South Africa, 9–13 November 2014. [Google Scholar]
- Hayden, F.G.; Whitley, R.J. Respiratory Syncytial Virus Antivirals: Problems and Progress. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hanke, L.; Perez, L.V.; Sheward, D.J.; Das, H.; Schulte, T.; Moliner-Morro, A.; Corcoran, M.; Achour, A.; Hedestam, G.B.K.; Hällberg, B.M.; et al. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Khanna, C.; Waldrep, J.C.; Anderson, P.M.; Weischelbaum, R.W.; Hasz, D.E.; Katsanis, E.; Klausner, J.S. Nebulized Interleukin 2 Liposomes: Aerosol Characteristics and Biodistribution. J. Pharm. Pharmacol. 1997, 49, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Anderson, P.M.; Hasz, D.E.; Katsanis, E.; Neville, M.; Klausner, J.S. Interleukin-2 liposome inhalation therapy is safe and effective for dogs with spontaneous pulmonary metastases. Cancer 1997, 79, 1409–1421. [Google Scholar] [CrossRef] [Green Version]
- Koussoroplis, S.J.; Paulissen, G.; Tyteca, D.; Goldansaz, H.; Todoroff, J.; Barilly, C.; Uyttenhove, C.; Van Snick, J.; Cataldo, D.D.; Vanbever, R. PEGylation of antibody fragments greatly increases their local residence time following delivery to the respiratory tract. J. Control. Release 2014, 187, 91–100. [Google Scholar] [CrossRef]
- Saluja, V.; Amorij, J.-P.; Kapteyn, J.C.; De Boer, A.; Frijlink, H.W.; Hinrichs, W.L.J. A comparison between spray drying and spray freeze drying to produce an influenza subunit vaccine powder for inhalation. J. Control. Release 2010, 144, 127–133. [Google Scholar] [CrossRef]
- Cantin, A.M.; Woods, D.E.; Cloutier, D.; Dufour, E.K.; Leduc, R. Polyethylene glycol conjugation at Cys 232 prolongs the half-life of α 1 proteinase inhibitor. Am. J. Respir. Cell Mol. Biol. 2002, 27, 659–665. [Google Scholar] [CrossRef]
- McLeod, V.M.; Chan, L.J.; Ryan, G.M.; Porter, C.J.; Kaminskas, L.M. Optimal PEGylation can Improve the Exposure of Interferon in the Lungs Following Pulmonary Administration. J. Pharm. Sci. 2015, 104, 1421–1430. [Google Scholar] [CrossRef]
- Guichard, M.-J.; Leal, T.; Vanbever, R. PEGylation, an approach for improving the pulmonary delivery of biopharmaceuticals. Curr. Opin. Colloid Interface Sci. 2017, 31, 43–50. [Google Scholar] [CrossRef]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Dumont, J.A.; Bitonti, A.J.; Clark, D.; Evans, S.; Pickford, M.; Newman, S.P. Delivery of an Erythropoietin-Fc Fusion Protein by Inhalation in Humans through an Immunoglobulin Transport Pathway. J. Aerosol Med. 2005, 18, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, S. Liquid formulation for antibody drugs. Biochim. Biophys. Acta 2014, 1844, 2041–2052. [Google Scholar] [CrossRef]
- Hertel, S.; Winter, G.; Friess, W. Protein stability in pulmonary drug delivery via nebulization. Adv. Drug Deliv. Rev. 2015, 93, 79–94. [Google Scholar] [CrossRef] [PubMed]
- McElroy, M.C.; Kirton, C.; Gliddon, D.; Wolff, R.K. Inhaled biopharmaceutical drug development: Nonclinical considerations and case studies. Inhal. Toxicol. 2013, 25, 219–232. [Google Scholar] [CrossRef]
- Bodier-Montagutelli, E.; Mayor, A.; Vecellio, L.; Respaud, R.; Heuzé-Vourc’h, N. Designing inhaled protein therapeutics for topical lung delivery: What are the next steps? Expert Opin. Drug Deliv. 2018, 15, 729–736. [Google Scholar] [CrossRef]
- Respaud, R.; Marchand, D.; Parent, C.; Pelat, T.; Thullier, P.; Tournamille, J.-F.; Viaud-Massuard, M.-C.; Diot, P.; Si-Tahar, M.; Vecellio, L.; et al. Effect of formulation on the stability and aerosol performance of a nebulized antibody. mAbs 2014, 6, 1347–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biddiscombe, M.F.; Usmani, O.S. Is there room for further innovation in inhaled therapy for airways disease? Breathe 2018, 14, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, G.S.; Chou, W.; Raimundo, K.; Trzaskoma, B.; Konstan, M.W. Randomized trial of efficacy and safety of dornase alfa delivered by eRapid nebulizer in cystic fibrosis patients. J. Cyst. Fibros. 2015, 14, 777–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geller, D.E.; Kesser, K.C. The I-neb Adaptive Aerosol Delivery System Enhances Delivery of α1-Antitrypsin with Controlled Inhalation. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, S55–S59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, P.; Schulte, M.; Wencker, M.; Herpich, C.H.; Klein, G.; Meyer, T. Lung deposition of inhaled α1-proteinase inhibitor in cystic fibrosis and α1-antitrypsin deficiency. Eur. Respir. J. 2009, 34, 354–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luisetti, M.; Kroneberg, P.; Suzuki, T.; Kadija, Z.; Muellinger, B.; Campo, I.; Gleske, J.; Rodi, G.; Zimlich, W.C.; Mariani, F.; et al. Physical properties, lung deposition modeling, and bioactivity of recombinant GM-CSF aerosolised with a highly efficient nebulizer. Pulm. Pharmacol. Ther. 2011, 24, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, E.; Volpi, S.; Salonini, E.; Van Der Wiel-Kooij, E.C.; Sintnicolaas, C.J.J.C.M.; Hop, W.C.J.; Assael, B.M.; Merkus, P.J.F.M.; Tiddens, H. Improved treatment response to dornase alfa in cystic fibrosis patients using controlled inhalation. Eur. Respir. J. 2011, 38, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Verma, R.; Garcia-Contreras, L. Inhalation drug delivery devices: Technology update. Med. Devices Evid. Res. 2015, 8, 131–139. [Google Scholar]
- Liao, Y.-H.; Brown, M.B.; Jones, S.A.; Nazir, T.; Martin, G.P. The effects of polyvinyl alcohol on the in vitro stability and delivery of spray-dried protein particles from surfactant-free HFA 134a-based pressurised metered dose inhalers. Int. J. Pharm. 2005, 304, 29–39. [Google Scholar] [CrossRef]
- Fathe, K.; Ferrati, S.; Moraga-Espinoza, D.; Yazdi, A.; Smyth, H.D.; Yazdi, A. Inhaled Biologics: From Preclinical to Product Approval. Curr. Pharm. Des. 2016, 22, 2501–2521. [Google Scholar] [CrossRef]
- Quinn, É.S.Á.; Forbes, R.T.; Williams, A.C.; Oliver, M.J.; McKenzie, L.; Purewal, T.S. Protein conformational stability in the hydrofluoroalkane propellants tetrafluoroethane and heptafluoropropane analysed by Fourier transform Raman spectroscopy. Int. J. Pharm. 1999, 186, 31–41. [Google Scholar] [CrossRef]
- Li, H.-Y.; Seville, P.C. Novel pMDI formulations for pulmonary delivery of proteins. Int. J. Pharm. 2010, 385, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Nyambura, B.K.; Kellaway, I.W.; Taylor, K.M. Insulin nanoparticles: Stability and aerosolization from pressurized metered dose inhalers. Int. J. Pharm. 2009, 375, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Yang, Z.; Peng, X.; Xin, F.; Xu, Y.; Feng, M.; Zhao, C.; Hu, H.; Wu, C. A novel bottom-up process to produce nanoparticles containing protein and peptide for suspension in hydrofluoroalkane propellants. Int. J. Pharm. 2011, 413, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Yang, Z.; Pan, X.; Chen, M.; Feng, M.; Wang, L.; Liu, H.; Shan, Z.; Wu, C. Stability and aerosolization of pressurized metered dose inhalers containing thymopentin nanoparticles produced using a bottom-up process. Int. J. Pharm. 2012, 427, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Rahimpour, Y.; Kouhsoltani, M.; Hamishehkar, H. Alternative carriers in dry powder inhaler formulations. Drug Discov. Today 2014, 19, 618–626. [Google Scholar] [CrossRef]
- White, S.; Bennett, D.B.; Cheu, S. Exubera®: Pharmaceutical Development of a Novel Product for Pulmonary Delivery of Insulin. Diabetes Technol. Ther. 2005, 7, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tsifansky, M.D.; Wu, C.-J.; Yang, H.I.; Schmidt, G.; Yeo, Y. Inhalable Antibiotic Delivery Using a Dry Powder Co-delivering Recombinant Deoxyribonuclease and Ciprofloxacin for Treatment of Cystic Fibrosis. Pharm. Res. 2010, 27, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Costantino, H.R.; Andya, J.D.; Nguyen, P.-A.; Dasovich, N.; Sweeney, T.D.; Shire, S.J.; Hsu, C.C.; Maa, Y.-F. Effect of Mannitol Crystallization on the Stability and Aerosol Performance of a Spray-Dried Pharmaceutical Protein, Recombinant Humanized anti-IgE Monoclonal Antibody. J. Pharm. Sci. 1998, 87, 1406–1411. [Google Scholar] [CrossRef]
- Schüle, S.; Schulzfademrecht, T.; Garidel, P.; Bechtoldpeters, K.; Friess, W. Stabilization of IgG1 in spray-dried powders for inhalation. Eur. J. Pharm. Biopharm. 2008, 69, 793–807. [Google Scholar] [CrossRef]
- Ramezani, V.; Vatanara, A.; Seyedabadi, M.; Meibodi, M.N.; Fanaei, H. Application of cyclodextrins in antibody microparticles: Potentials for antibody protection in spray drying. Drug Dev. Ind. Pharm. 2017, 43, 1103–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensink, M.A.; Frijlink, H.W.; Maarschalk, K.V.D.V.; Hinrichs, W.L.J. How sugars protect proteins in the solid state and during drying (review): Mechanisms of stabilization in relation to stress conditions. Eur. J. Pharm. Biopharm. 2017, 114, 288–295. [Google Scholar] [CrossRef]
- Quarta, E.; Chierici, V.; Flammini, L.; Tognolini, M.; Barocelli, E.; Cantoni, A.M.; Dujovny, G.; Probst, S.E.; Sonvico, F.; Colombo, G.; et al. Excipient-free pulmonary insulin dry powder: Pharmacokinetic and pharmacodynamics profiles in rats. J. Control. Release 2020, 323, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; MacKenzie, B.; Koleng, J.J.; Maier, E.; Warnken, Z.N.; Williams, R.O. Development of an Excipient-Free Peptide Dry Powder Inhalation for the Treatment of Pulmonary Fibrosis. Mol. Pharm. 2020, 17, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Wanning, S.; Süverkrüp, R.; Lamprecht, A. Pharmaceutical spray freeze drying. Int. J. Pharm. 2015, 488, 136–153. [Google Scholar] [CrossRef]
- Mack, P.; Horvath, K.; Garcia, A.; Tully, J.; Maynor, B. Particle engineering for inhalation formulation and delivery of biotherapeutics. Inhalation 2012, 6, 6–20. [Google Scholar]
- Peeters, B.; Tonnis, W.F.; Murugappan, S.; Rottier, P.; Koch, G.; Frijlink, H.; Huckriede, A.; Hinrichs, W.L.J. Pulmonary immunization of chickens using non-adjuvanted spray-freeze dried whole inactivated virus vaccine completely protects against highly pathogenic H5N1 avian influenza virus. Vaccine 2014, 32, 6445–6450. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.C.; Boss, A.H. Technosphere insulin technology. Diabetes Technol. Ther. 2007, 9 (Suppl. 1), S65–72. [Google Scholar] [CrossRef] [PubMed]
- Heise, T.; Brugger, A.; Cook, C.; Eckers, U.; Hutchcraft, A.; Nosek, L.; Rave, K.; Troeger, J.; Valaitis, P.; White, S.; et al. Promaxx inhaled insulin: Safe and efficacious administration with a commercially available dry powder inhaler. Diabetes Obes. Metab. 2009, 11, 455–459. [Google Scholar] [CrossRef]
- Wilson, E.M.; Luft, J.C.; DeSimone, J.M. Formulation of High-Performance Dry Powder Aerosols for Pulmonary Protein Delivery. Pharm. Res. 2018, 35, 195. [Google Scholar] [CrossRef]
- De Boer, A.H.; Hagedoorn, P.; Hoppentocht, M.; Buttini, F.; Grasmeijer, F.; Frijlink, H.W. Dry powder inhalation: Past, present and future. Expert Opin. Drug Deliv. 2017, 14, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Al-Tabakha, M.M. Future prospect of insulin inhalation for diabetic patients: The case of Afrezza versus Exubera. J. Control. Release 2015, 215, 25–38. [Google Scholar] [CrossRef]
- Colombo, R.E.; Fiorentino, C.; Dodd, L.E.; Hunsberger, S.; Haney, C.; Barrett, K.; Nabha, L.; Davey, R.T., Jr.; Olivier, K.N. A phase 1 randomized, double-blind, placebo-controlled, crossover trial of DAS181 (Fludase®) in adult subjects with well-controlled asthma. BMC Infect. Dis. 2015, 16, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerem, E.; Blau, H.; Shteinberg, M.; Efrati, O.; Alon, S.; Dekel, E.; Amit, B.; Raul, C.; Brill-Almon, E.; Fux, L.; et al. WS01.2 Phase II clinical trial results of alidornase alfa for the treatment of cystic fibrosis. J. Cyst. Fibros. 2017, 16, S1–S62. [Google Scholar] [CrossRef]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Sorrells, S.; Camprubí, S.; Griffin, R.; Chen, J.; Ayguasanosa, J. SPARTA clinical trial design: Exploring the efficacy and safety of two dose regimens of alpha1-proteinase inhibitor augmentation therapy in alpha1-antitrypsin deficiency. Respir. Med. 2015, 109, 490–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggar, A.; Chen, J.; Chmiel, J.F.; Dorkin, H.L.; Flume, P.A.; Griffin, R.; Nichols, D.; Donaldson, S.H. Inhaled alpha 1 -proteinase inhibitor therapy in patients with cystic fibrosis. J. Cyst. Fibros. 2016, 15, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, H.J.; Borowitz, D.; Christiansen, D.H.; Morris, E.M.; Nash, M.L.; Ramsey, B.W.; Rosenstein, B.J.; Smith, A.L.; Wohl, M.E. Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis. N. Engl. J. Med. 1994, 331, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Bauer, S.; Strauss, P.; Jaffe, N.; Armoni, S.; Pugatsch, T.; Shoseyov, D.; Tov, N. Safety and Efficacy of Inhaled Human Alpha-1 Antitrypsin (AAT) in Cystic Fibrosis (CF): A Report of a Phase II Clinical Study. Am. J. Respir. Crit. Care Med. 2009, 179, A1185. [Google Scholar]
- Brantly, M.; Stocks, J.; Rouhani, F.; Lascano, J.; Jeffers, A.; Nolte, J.; Owens, S.; Tucker, T.A.; Tov, N. Inhaled Alpha-1-Antitrypsin Restores Lower Respiratory Tract Protease-Anti-Protease Homeostasis And Reduces Inflammation In Alpha-1 Antitrypsin Deficient Individuals: A Phase 2 Clinical Study Using Inhaled Kamada-Api. Am. J. Respir. Crit. Care Med. 2017, 195, A7677. [Google Scholar]
- Wenzel, S. Asthma phenotypes: The evolution from clinical to molecular approaches. Nat. Med. 2012, 18, 716. [Google Scholar] [CrossRef]
- Wenzel, S.; Wilbraham, D.; Fuller, R.; Getz, E.B.; Longphre, M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: Results of two phase 2a studies. Lancet 2007, 370, 1422–1431. [Google Scholar] [CrossRef]
- Matschiner, G.; Huang, S.; Constant, S.; Rattenstetter, B.; Gille, H.; Hohlbaum, A.M.; Koller, B.; Keeling, D.; Fitzgerald, M. The Discovery and Development of AZD1402/PRS-060, an Inhaled, Potent and Selective Antagonist of the IL-4 Receptor Alpha. Airway Pharmacol. Treat. 2018, 52, PA1047. [Google Scholar]
- Bruns, I.; Fitzgerald, M.F.; Pardali, K.; Gardiner, P.; Keeling, D.; Axelsson, L.; Jiang, F.; Lickliter, J.; Close, D. First-in-human data for the inhaled IL-4Rα antagonist AZD1402/PRS-060 reveals a promising clinical profile for the treatment of asthma. Am. J. Respir. Crit. Care Med. 2019, 199, A7476. [Google Scholar]
- Chan, R.W.Y.; Chan, M.C.-W.; Wong, A.C.N.; Karamanska, R.; Dell, A.; Haslam, S.M.; Sihoe, A.D.L.; Chui, W.H.; Triana-Baltzer, G.; Li, Q.; et al. DAS181 Inhibits H5N1 Influenza Virus Infection of Human Lung Tissues. Antimicrob. Agents Chemother. 2009, 53, 3935–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, R.B.; Hansen, C.; Sanders, R.L.; Hawley, S.; Li, T.; Steigbigel, R.T. A Phase II Study of DAS181, a Novel Host Directed Antiviral for the Treatment of Influenza Infection. J. Infect. Dis. 2012, 206, 1844–1851. [Google Scholar] [CrossRef] [Green Version]
- Boxall, C.; Dudley, S.; Beegan, R.; Tear, V.; Hrebien, S.; Lunn, K.; Monk, P. Effect of inhaled sng001 (interferon-beta 1a) on sputum and blood antiviral biomarkers following a respiratory virus infection in asthmatic subjects. Eur. Respir. Soc. 2013, 42, 3182. [Google Scholar]
- Monk, P.; Reynolds, S.; Lunn, K.; Beegan, R.; Roberts, J.; Tear, V. Upregulation of Antiviral Biomarkers in Sputum Cells Following Administration of Inhaled Interferon Beta to Patients with COPD. In Advances in COPD Pathogenesis; American Thoracic Society: New York, NY, USA, 2019; p. A4045. [Google Scholar]
- Djukanović, R.; Harrison, T.; Johnston, S.L.; Gabbay, F.; Wark, P.; Thomson, N.C.; Niven, R.; Singh, D.; Reddel, H.K.; Davies, D.E.; et al. The Effect of Inhaled IFN-β on Worsening of Asthma Symptoms Caused by Viral Infections. A Randomized Trial. Am. J. Respir. Crit. Care Med. 2014, 190, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Majedi, S.; Majedi, S. Existing drugs as treatment options for COVID-19: A brief survey of some recent results. J. Chem. Lett. 2020, 1, 2–8. [Google Scholar]
- Synairgen Announces Positive Results from Trial of SNG001 in Hospitalised COVID-19 Patients. 2020. Available online: https://www.globenewswire.com/news-release/2020/07/20/2064154/0/en/Synairgen-announces-positive-results-from-trial-of-SNG001-in-hospitalised-COVID-19-patients.html (accessed on 8 October 2020).
- Enk, A.H.; Nashan, D.; Rübben, A.; Knop, J. High dose inhalation interleukin-2 therapy for lung metastases in patients with malignant melanoma. Cancer 2000, 88, 2042–2046. [Google Scholar] [CrossRef]
- Tunney, M.M.; Payne, J.E.; McGrath, S.J.; Einarsson, G.G.; Ingram, R.J.; Gilpin, D.F.; Juarez-Perez, V.; Elborn, J.S. Activity of hypothiocyanite and lactoferrin (ALX-009) against respiratory cystic fibrosis pathogens in sputum. J. Antimicrob. Chemother. 2018, 73, 3391–3397. [Google Scholar] [CrossRef] [PubMed]
- Stringer, K.A.; Schumacher, K.R.; Caruthers, R.L.; Nasr, S.Z.; Myers, J.L. Study Design: Phase II safety and efficacy of inhaled tissue plasminogen activator (activase) for the treatment of acute pediatric plastic bronchitis. In Bronchopulmonary Dysplasia; American Thoracic Society: New York, NY, USA, 2017; p. A2179. [Google Scholar]
- McGill, J.B.; Ahn, D.; Edelman, S.V.; Kilpatrick, C.R.; Cavaiola, T.S. Making Insulin Accessible: Does Inhaled Insulin Fill an Unmet Need? Adv. Ther. 2016, 33, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Mitri, J.; Pittas, A.G. Inhaled insulin—What went wrong. Nat. Clin. Pract. Endocrinol. Metab. 2009, 5, 24–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kammona, O.; Kiparissides, C. Recent advances in nanocarrier-based mucosal delivery of biomolecules. J. Control. Release 2012, 161, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, R.R.; Das, S. Inhaled Insulin—Current Direction of Insulin Research. J. Clin. Diagn. Res. 2017, 11, OE01–OE02. [Google Scholar] [CrossRef] [PubMed]
- Rochman, Y.; Leonard, W.J. Thymic stromal lymphopoietin: A new cytokine in asthma. Curr. Opin. Pharmacol. 2008, 8, 249–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauvreau, G.; Hohlfeld, J.; Grant, S.; Jain, M.; Cabanski, M.; Pertel, P.; Boulet, L.-P.; Cockcroft, D.; Davis, B.; Fitzgerald, J.; et al. Efficacy and safety of an inhaled anti-TSLP antibody fragment in adults with mild atopic asthma. Am. J. Respir. Crit. Care Med. 2020, A4207. [Google Scholar]
- Awwad, S.; Angkawinitwong, U. Overview of Antibody Drug Delivery. Pharmaceutics 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duggan, S. Caplacizumab: First Global Approval. Drugs 2018, 78, 1639–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Results from the First-in-Infant Phase I/IIa Study with the Anti-RSV Nanobody, ALX-0171. Available online: https://www.ablynx.com/uploads/data/files/ablynx_alx-0171_first-ininfant%20study%20results_webcast%20presentation.pdf (accessed on 20 October 2020).
- Löwensteyn, Y.N.; Bont, L.J. Clinical Development of Respiratory Syncytial Virus Antivirals—What We Can Learn From Oseltamivir. J. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Konwarh, R. Nanobodies: Prospects of Expanding the Gamut of Neutralizing Antibodies Against the Novel Coronavirus, SARS-CoV-2. Front. Immunol. 2020, 11, 1531. [Google Scholar] [CrossRef]
- Klonoff, D.C. Afrezza inhaled insulin: The fastest-acting FDA-approved insulin on the market has favorable properties. J. Diabetes Sci. Technol. 2014, 8, 1071–1073. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, J.P.; Amin, N.; Marino, M.; Gotfried, M.; Meyer, T.; Sommerer, K.; Baughman, R.A. Insulin Lung Deposition and Clearance Following Technosphere® Insulin Inhalation Powder Administration. Pharm. Res. 2011, 28, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- McCoy, K.; Hamilton, S.; Johnson, C. Effects of 12-Week Administration of Dornase Alfa in Patients with Advanced Cystic Fibrosis Lung Disease. Chest 1996, 110, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Hollander, P.A.; Blonde, L.; Rowe, R.; Mehta, A.E.; Milburn, J.L.; Hershon, K.S.; Chiasson, J.-L.; Levin, S.R. Efficacy and safety of inhaled insulin (exubera) compared with subcutaneous insulin therapy in patients with type 2 diabetes: Results of a 6-month, randomized, comparative trial. Diabetes Care 2004, 27, 2356–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, B.G.; Cortez, J.; Kolli, M.; Sharma, A.; Delaney-Black, V.; Chen, X. Aerosolized surfactant in neonatal respiratory distress syndrome: Phase I study. Early Hum. Dev. 2019, 134, 19–25. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monoclonal Antibody | Brand Name | Target | Company | Approved Respiratory Indication(s) | Year of Initial Approval |
|---|---|---|---|---|---|
| Rituximab | Rituxan® | CD20 | Genentech | GPA; MPA | 1997 |
| Truxima® | Celltrion | 2018 | |||
| Ruxience™ | Pfizer | 2019 | |||
| Palivizumab | Synagis® | RSV envelope Fusion protein | AstraZeneca/AbbVie | Prophylaxis of RSV-associated serious lower respiratory tract disease | 1998 |
| Omalizumab | Xolair® | IgE | Genentech/Novartis | Moderate-to-severe persistent asthma | 2003 |
| Bevacizumab | Avastin® | VEGF | Genentech | Non-squamous NSCLC | 2004 |
| Mvasi™ | Amgen | 2017 | |||
| Zirabev™ | Pfizer | 2019 | |||
| Ipilimumab | Yervoy® | CTLA-4 | BMS | NSCLC | 2011 |
| Raxibacumab | N.A. | B. anthracis PA | Emergent BioSolutions | Inhalational anthrax | 2012 |
| Nivolumab | Opdivo® | PD-1 | BMS | NSCLC; SCLC | 2014 |
| Pembrolizumab | Keytruda® | PD-1 | MSD | NSCLC; SCLC | 2014 |
| Ramucirumab | Cyramza® | VEGFR-2 | Eli Lilly | Metastatic NSCLC | 2014 |
| Mepolizumab | Nucala® | IL-5 | GSK | SEA, EGPA | 2015 |
| Necitumumab | Portrazza® | EGFR | Eli Lilly | Metastatic squamous NSCLC | 2015 |
| Atezolizumab | Tecentriq® | PD-L1 | Genentech | NSCLC; SCLC | 2016 |
| Obiltoxaximab | Anthim® | B. anthracis PA | Elusys Therapeutics | Inhalational anthrax | 2016 |
| Reslizumab | Cinqair® | IL-5 | Teva | SEA | 2016 |
| Benralizumab | Fasenra™ | IL-5Rα | AstraZeneca | SEA | 2017 |
| Dupilumab | Dupixent® | IL-4Rα | Regeneron/Sanofi | Moderate-to-severe asthma, CRSwNP | 2018 |
| Durvalumab | Imfinzi® | PD-L1 | AstraZeneca | NSCLC; SCLC | 2017 |
| Characteristics | Biological Drugs | Small-Molecule Drugs |
|---|---|---|
| Molecular weight | >1 kDa, often much larger | Usually <700 Da |
| Molecular composition and structure | Proteins with known amino acid sequence, higher order structure, and diverse post-translational modifications | Organic chemicals with well-defined structure |
| Mechanisms of action | Fab-dependent binding to target epitope; Fc-dependent complement activation and effector cell recruitment | Typically enzyme or receptor inhibition |
| Pharmacological target | Normally extracellular | Extracellular or intracellular |
| Stability | Sensitive to physical conditions (e.g., temperature changes, shear stresses, pH, light) and proteases | Comparatively stable |
| Routes of administration | Mostly parenteral | Wide-ranging; oral route feasible if desired |
| In vivo half-life | Generally long; dosing intervals in weeks or months possible | Generally short; once-daily and more frequent dosing common |
| Specificity | High | Variable |
| In vivo degradation | Proteolytic metabolism | Enzymatic metabolism by cytochrome P450 |
| Manufacturing process | Recombinant DNA technology; expansion in a unique cell line; purification from growth media | Chemical synthesis |
| Price | High | Variable |
| Me-too | Biosimilar (highly similar to a reference product) | Generic (identical entity) |
| Drug | Target Disease | Formulation | Animal | Target/Format | Administration/Device | Reference & Year |
|---|---|---|---|---|---|---|
| Aldesleukin | Pulmonary metastases | Liposome | Dogs | IL-2 | Puritan Bennet™ twin-jet nebuliser | [109,110] 1997 |
| ALX-0171 | RSV infection | Inhaled solution | Cotton rats | Anti-Fusion protein trivalent Nanobody® | AKITA2 APIXNEB® nebuliser | [27,96] 2016 |
| ALX-0171 | RSV infection | Inhaled solution | New born lambs | Anti-Fusion protein trivalent Nanobody® | Aeroneb® Solo system | [97] 2018 |
| Anti-IL-17A PEG40-F(ab′)2 and Anti-IL-13 PEG40-Fab’ | Asthma | Inhaled solution | NMRI mice | Anti-IL-17A F(ab’)2 and Anti-IL-13 Fab | Intranasal instillation | [111] 2014 |
| Anti-IL-17A PEG40-Fab’ | Asthma | Inhaled solution | Mice, rats and rabbits | Anti-IL-17A Fab | Intratracheal instillation | [37] 2017 |
| Anti-IL-17A PEG20-Fab’, Anti-IL-17A PEG40-Fab’ and Anti-IL-13 PEG40-Fab’ | Asthma | Inhaled solution | Mice | Anti-IL-17A and anti-IL-13 Fab | Intratracheal instillation | [60] 2018 |
| Cetuximab | Lung tumour | Inhaled solution | Balb/c nude mice | Anti-EGFR mAb | Aeroneb Pro™ mesh nebuliser | [22,33] 2011 |
| Cetuximab | Lung tumour | Inhaled solution | Balb/c nude mice and cynomolgus macaques | Anti-EGFR mAb | Microsprayer® IA-1b aerosoliser | [21] 2014 |
| CA154_582 | Asthma | Inhaled solution | Balb/c mice | Anti-IL-13 Fab | inExpose nebulisation system | [88] 2012 |
| CDP7766 | Asthma | Inhaled solution | Cynomolgus macaques | Anti-IL-13 Fab | PARI eFlow® mesh nebuliser | [89] 2017 |
| EpoFc | Anemia | Inhaled solution | Cynomolgus monkeys | Erythropoietin Fc-fusion protein | Aeroneb Pro® nebuliser | [57] 2004 |
| FSHFc | Infertility | Inhaled solution | Cynomolgus monkeys | FSH Fc-fusion protein | Aeroneb Pro™ nebuliser | [81] 2005 |
| GSK1995057 | Acute lung injury | Inhaled solution | Cynomolgus monkeys | Anti-TNF receptor-1 dAb | Intratracheal instillation | [38] 2018 |
| hGH | Growth hormone deficiency | SD powder | Wistar rats | hGH | Dry Powder Insufflator™ | [65] 2004 |
| Influenza subunit vaccine | Influenza | SD and SFD powder | Balb/c mice | Surface glycoprotein haemagglutinin | Dry Powder Insufflator™ | [112] 2010 |
| IFNβFc | Multiple sclerosis | Inhaled solution | Cynomolgus monkeys | IFNβ Fc-fusion protein | Aeroneb Pro® mesh nebuliser | [82] 2012 |
| IgG 43RCA-G1 | Ricin intoxication | Inhaled solution | Balb/c mice/cynomolgus macaques | Anti-ricin mAb derived from scFv 43RCA | Micropipette tip and Aerogen® Solo mesh nebuliser | [36] 2016 |
| Infliximab | Asthma | SD powder | Balb/c mice | Anti-TNFα mAb | Dry Powder Insufflator™ | [34] 2019 |
| p55-specific dAb | Ventilator-induced lung injury | Inhaled solution | C57BL6 mice | Anti-p55 TNF receptor dAb | Intratracheal instillation | [91] 2012 |
| PEG-rhα1-PI | Hereditary emphysema | Solution | CD1 mice | α1-PI | Intranasal instillation | [113] 2002 |
| PEG12-IFNαPEG40-IFNα | Cancer or fibrosis | Inhaled solution | SD rats | PEGylated IFNα | Intratracheal instillation | [114] 2014 |
| Name | API | Development Stage | Company/Sponsor | Clinical Application | Delivery Device | Formulation | Reference, Year & Clinical Trial Number |
|---|---|---|---|---|---|---|---|
| Inhaled proteins | |||||||
| Afrezza® | Insulin, 5.7 kDa | Marketed in 2014 | MannKind | Diabetes mellitus | Dreamboat® inhaler | Technosphere® insulin inhalation powder | [187,188] 2014 |
| Alpha-1 HC | Human α1-PI, 52 kDa | Phase II | Grifols Therapeutics | CF | AKITA2 APIXNEB® nebuliser system | Inhaled solution | [158] 2016, NCT01684410 |
| AZD1402/PRS-060 | IL4 mutein (IL-4Rα antagonist), ~18 kDa | Phase I | AstraZeneca & Pieris Pharmaceuticals | Asthma | InnoSpire Go mesh nebuliser | Inhaled solution | [164,165] 2019, NCT03384290 and NCT03574805 |
| Alteplase | rt-PA, 70 kDa | Phase II | University of Michigan & Genentech | Acute plastic Bronchitis | Nebuliser | Inhaled solution | [175] 2017, NCT02315898 |
| ALX-009 | OSCN-/bLF, 80 kDa | Phase I | Alaxia SAS | P. aeruginosa and Bcc infection in CF | Nebuliser | Inhaled solution | [174] 2018, NCT02598999 |
| Alidornase alfa (PRX-110, AIR Dnase™) | rhDNase I, 37 kDa | Phase I | Protalix | CF | Philips Respironics I-neb AAD inhaler system | Inhaled solution | [155] 2017, NCT02605590 |
| Dornase alfa (Pulmozyme®) | rhDNase I, 37 kDa | Marketed in 1993 | Genentech | CF | Jet nebuliser/air compressor combinations | Inhaled solution | [189] 1996 |
| Dornase alfa | rhDNase I, 37 kDa | Phase IV | Erasmus Medical Centre-Sophia Children’s Hospital | CF | AKITA2 APIXNEB® nebuliser system | Inhaled solution | [128] 2011 |
| Dornase alfa | rhDNase I, 37 kDa | Phase IV | PARI | CF | eRapid™ nebuliser system | Inhaled solution | [124] 2015, NCT01712334 |
| DAS181 (Fludase®) | Recombinant sialidase fusion protein, 46 kDa | Phase I/II | Ansun BioPharma | Parainfluenza infection | Cyclohaler® DPI | Dry powder | [154,167] 2015, NCT01037205, NCT01924793 NCT01113034 |
| Compassionate use/Phase III | Renmin Hospital of Wuhan University & Ansun BioPharma | COVID-19 | Nebuliser | Inhaled solution | NCT04324489 NCT03808922 | ||
| Exubera® | Insulin, 5.7 kDa | Marketed in 2006; withdrawn in 2007 | Pfizer/Nektar Therapeutics | Diabetes mellitus | Exubera® DPI inhaler | SD powder | [190] 2004 |
| EpoFc | Epo Fc-fusion protein | Phase I | Syntonix Pharmaceuticals | Anemia | Aeroneb® Pro nebuliser | Inhaled solution | [117] 2005 |
| GM-CSF (Leukine®, Sargramostim) | rhuGM-CSF, 14 kDa | Phase I | Milton S. Hershey Medical Center | RASP | Nebuliser | Inhaled solution | NCT02601365 |
| Phase II | Children’s Hospital Medical Center, Cincinnati | PAP | Nebuliser | Inhaled solution | NCT01511068 | ||
| Phase II | Peking Union Medical College Hospital | PAP | Nebuliser | Inhaled solution | NCT02243228 | ||
| Kamada AAT | Alpha-1-antitrypsin, 52 kDa | Phase II | Kamada | CF | PARI eFlow® nebuliser | Inhaled solution | [160] 2009 |
| Kamada-API | Alpha-1-antitrypsin, 52 kDa | Phase II/III | Kamada | AATD | PARI eFlow® nebuliser | Inhaled solution | [161] 2017, NCT04204252 |
| Pitrakinra (Aerovant®, AER-001) | IL4 mutein (IL-4Rα antagonist), 15 kDa | Phase II | Bayer | Asthma | PARI LC Plus nebuliser | Inhaled solution | [163] 2007, NCT00535031 |
| Aldesleukin (Proleukin®) | Recombinant IL-2, 15 kDa | Phase I/II | M.D. Anderson Cancer Center | Lung metastases | Nebuliser | Inhaled solution | [173] 2000, NCT01590069 |
| Survanta® | Bovine surfactant (phospholipids, triglycerides, free fatty acids and proteins, etc.) | Phase I/II | DMC Foundation | RDS | MiniHeart jet nebuliser | Inhaled solution | [191] 2019, NCT02294630 |
| SNG001 | Interferon-beta 1a, 22 kDa | Phase II | Synairgen | RVI in asthma | Nebuliser | Inhaled solution | [168,170] 2014, NCT01126177 |
| Phase II | Synairgen | COPD | Nebuliser | Inhaled solution | [169] 2019 | ||
| Phase II | Synairgen | COVID-19 | Nebuliser | Inhaled solution | [171] 2020, NCT04385095 | ||
| Inhaled mAbs and mAb fragments | |||||||
| ALX-0171 | Anti-F protein trivalent Nanobody®, 42 kDa | Phase II | Ablynx | RSV infection | FOX-Flamingo inhalation system | Inhaled solution | 2017, NCT02979431 and NCT03418571 |
| CSJ117 | Anti-TSLP antibody fragment, 46 kDa | Phase I/II | Novartis | Asthma | Concept1 device (single dose DPI) | PulmoSol™ engineered powder | [181] 2020, NCT03138811 and NCT04410523 |
| E25 | Omalizumab, 149 kDa | Phase III | Genentech/Novartis | Asthma | PARI IS-2 nebuliser | Inhaled solution | [35] 1999 |
| GSK1995057 | Anti-TNFR1 dAb, 13 kDa | Phase I | GSK | Acute lung injury | PARI eFlow® nebuliser | Inhaled solution | [38] 2018, NCT01587807 |
| GSK2862277 | Anti-TNFR1 dAb, 13 kDa | Phase II | GSK | Postoperative lung injury | PARI eFlow® nebuliser | Inhaled solution | [93] 2020, NCT02221037 |
| VR942/ CDP7766 | Anti-IL-13 mAb fragment | Phase I | UCB Pharma | Asthma | Multidose F1P DPI | Dry powder | [45] 2018, NCT02473939 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, W.; Pan, H.W.; Vllasaliu, D.; Lam, J.K.W. Pulmonary Delivery of Biological Drugs. Pharmaceutics 2020, 12, 1025. https://doi.org/10.3390/pharmaceutics12111025
Liang W, Pan HW, Vllasaliu D, Lam JKW. Pulmonary Delivery of Biological Drugs. Pharmaceutics. 2020; 12(11):1025. https://doi.org/10.3390/pharmaceutics12111025
Chicago/Turabian StyleLiang, Wanling, Harry W. Pan, Driton Vllasaliu, and Jenny K. W. Lam. 2020. "Pulmonary Delivery of Biological Drugs" Pharmaceutics 12, no. 11: 1025. https://doi.org/10.3390/pharmaceutics12111025
APA StyleLiang, W., Pan, H. W., Vllasaliu, D., & Lam, J. K. W. (2020). Pulmonary Delivery of Biological Drugs. Pharmaceutics, 12(11), 1025. https://doi.org/10.3390/pharmaceutics12111025