Influence of Probenecid on the Pharmacokinetics and Pharmacodynamics of Sorafenib

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Study Procedures
2.3. Pharmacokinetic Sampling
2.4. Skin Biopsies
2.5. In Vitro Transport Assay
2.6. Toxicity
2.7. Statistical Analysis
3. Results
3.1. Patient Characteristics
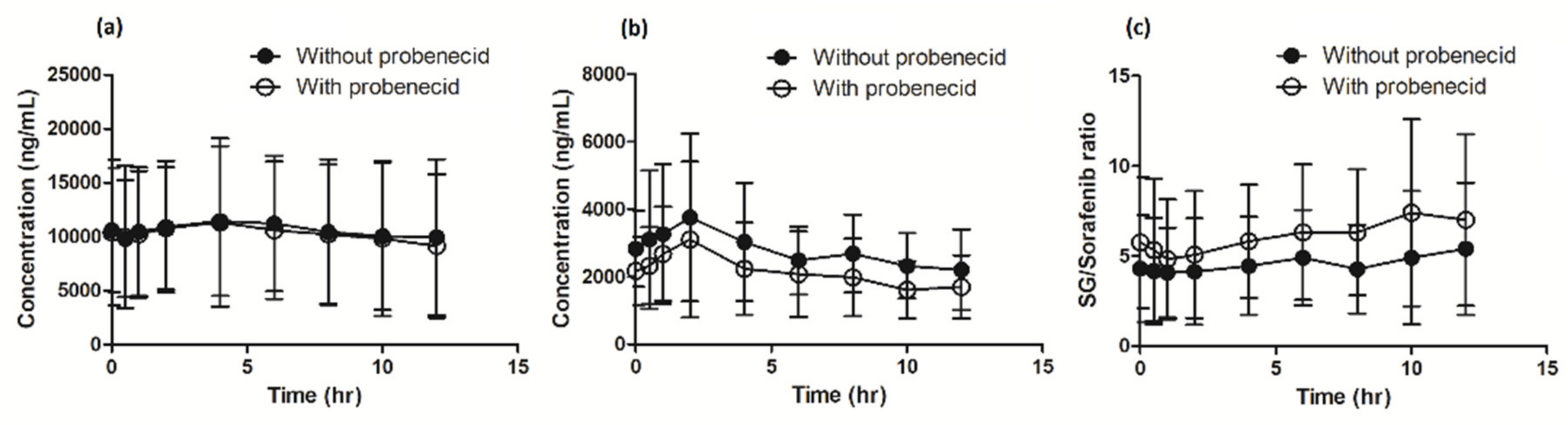
3.2. Pharmacokinetics
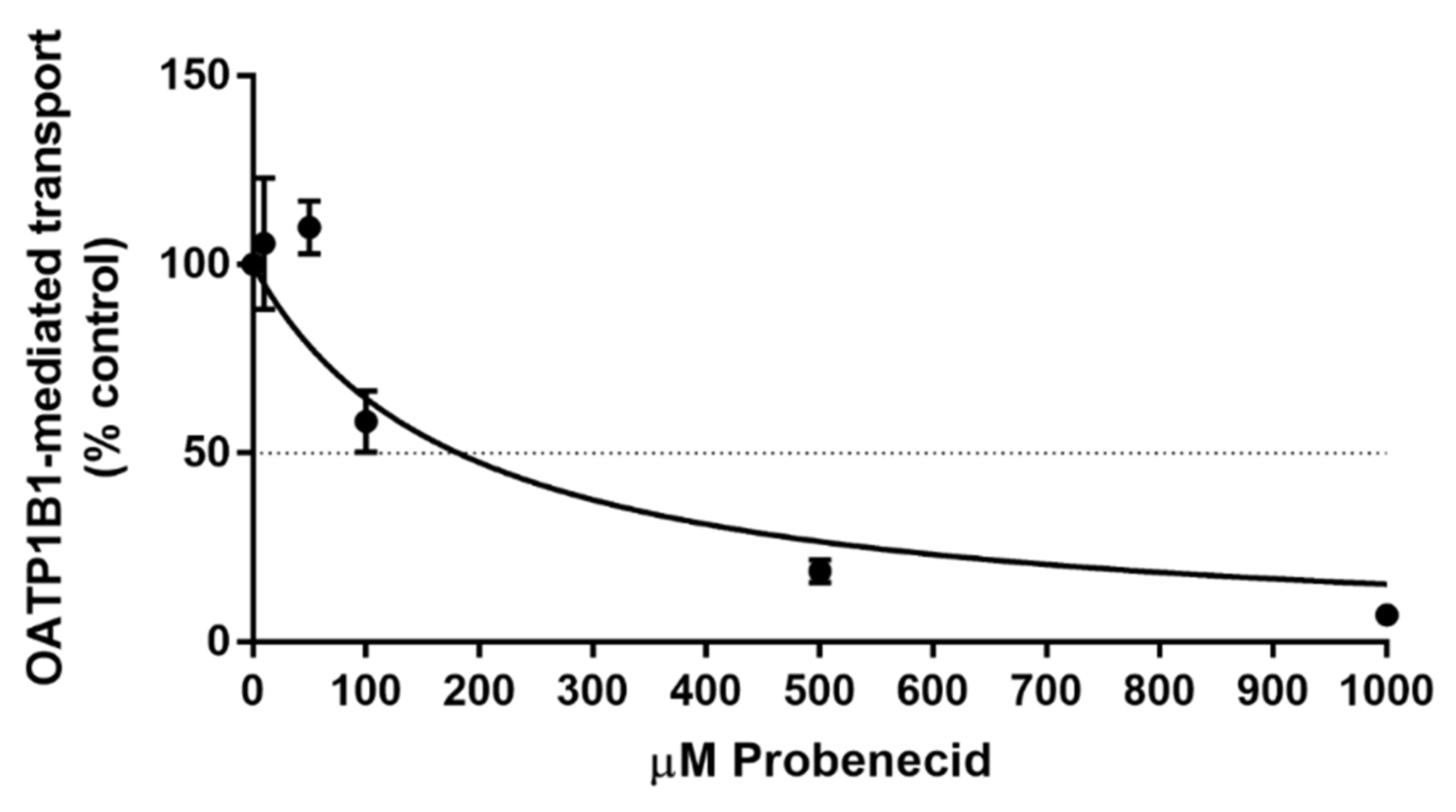
3.3. In Vitro Transport Assay
3.4. Toxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hartmann, J.T.; Haap, M.; Kopp, H.G.; Lipp, H.P. Tyrosine kinase inhibitors-a review on pharmacology, metabolism and side effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef]
- Anderson, R.T.; Keating, K.N.; Doll, H.A.; Camacho, F. The Hand-Foot Skin Reaction and Quality of Life Questionnaire: An Assessment Tool for Oncology. Oncologist 2015, 20, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Shen, Y.C.; Cheng, A.L. Sorafenib for the treatment of hepatocellular carcinoma across geographic regions. Expert Rev. Clin. Pharmacol. 2009, 2, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gu, J. Hand-foot skin reaction with vascular endothelial growth factor receptor tyrosine kinase inhibitors in cancer patients: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2017, 119, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rolland, F.; et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar] [CrossRef]
- Rollig, C.; Serve, H.; Hüttmann, A.; Noppeney, R.; Müller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Santos, F.P.S.; Kantarjian, H.; Fava, C.; O’Brien, S.; Garcia-Manero, G.; Ravandi, F.; Wierda, W.; Thomas, D.; Shan, J.; Cortes, J. Clinical impact of dose reductions and interruptions of second-generation tyrosine kinase inhibitors in patients with chronic myeloid leukaemia. Br. J. Haematol. 2010, 150, 303–312. [Google Scholar] [CrossRef]
- Thomas, L.; Lai, S.Y.; Dong, W.; Feng, L.; Dadu, R.; Regone, R.M.; Cabanillas, M.E. Sorafenib in metastatic thyroid cancer: A systematic review. Oncologist 2014, 19, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, E.I.; Gibson, A.A.; Hu, S.; Vasilyeva, A.; Orwick, S.J.; Du, G.; Mascara, G.P.; Ong, S.S.; Chen, T.; Vogel, P.; et al. Multikinase Inhibitors Induce Cutaneous Toxicity through OAT6-Mediated Uptake and MAP3K7-Driven Cell Death. Cancer Res. 2016, 76, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overbosch, D.; Van Gulpen, C.; Hermans, J.; Mattie, H. The effect of probenecid on the renal tubular excretion of benzylpenicillin. Br. J. Clin. Pharmacol. 1988, 25, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bins, S.; van Doorn, L.; Phelps, M.A.; Gibson, A.A.; Hu, H.; Li, L.; Vasilyeva, A.; Du, G.; Hamberg, P.; Eskens, F.; et al. Influence of OATP1B1 Function on the Disposition of Sorafenib-beta-D-Glucuronide. Clin. Transl. Sci. 2017, 10, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendonza, A.; Hanna, I.; Meyers, D.; Koo, P.; Neelakantham, S.; Zhu, B.; Majumdar, T.; Rebello, S.; Sunkara, G.; Chen, J. Assessment of pharmacokinetic drug-drug interaction between pradigastat and atazanavir or probenecid. J. Clin. Pharmacol. 2016, 56, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Uchaipichat, V.; Mackenzie, P.I.; Guo, X.-H.; Gardner-Stephen, D.; Galetin, A.; Houston, J.B.; Miners, J.O. Human udp-glucuronosyltransferases: Isoform selectivity and kinetics of 4-methylumbelliferone and 1-naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug Metab. Dispos. 2004, 32, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Hagos, F.T.; Daood, M.J.; Ocque, J.A.; Nolin, T.D.; Bayir, H.; Poloyac, S.M.; Kochanek, P.M.; Clark, R.S.; Empey, P.E. Probenecid, an organic anion transporter 1 and 3 inhibitor, increases plasma and brain exposure of N-acetylcysteine. Xenobiotica 2017, 47, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Minami, H.; Kawada, K.; Ebi, H.; Kitagawa, K.; Kim, Y.-I.; Araki, K.; Mukai, H.; Tahara, M.; Nakajima, H.; Nakajima, K. Phase I and pharmacokinetic study of sorafenib, an oral multikinase inhibitor, in Japanese patients with advanced refractory solid tumors. Cancer Sci. 2008, 99, 1492–1498. [Google Scholar] [CrossRef]
- Li, L.; Zhao, M.; Navid, F.; Pratz, K.; Smith, B.D.; Rudek, M.A.; Baker, S.D. Quantitation of sorafenib and its active metabolite sorafenib N-oxide in human plasma by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 3033–3038. [Google Scholar] [CrossRef] [Green Version]
- Lipworth, A.D.; Robert, C.; Zhu, A.X. Hand-foot syndrome (hand-foot skin reaction, palmar-plantar erythrodysesthesia): Focus on sorafenib and sunitinib. Oncology 2009, 77, 257–271. [Google Scholar] [CrossRef]
- Zimmerman, E.I.; Hu, S.; Roberts, J.L.; Gibson, A.A.; Orwick, S.J.; Li, L.; Sparreboom, A.; Baker, S.D. Contribution of OATP1B1 and OATP1B3 to the disposition of sorafenib and sorafenib-glucuronide. Clin. Cancer Res. 2013, 19, 1458–1466. [Google Scholar] [CrossRef] [Green Version]
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. Localization and genomic organization of a new hepatocellular organic anion transporting polypeptide. J. Biol. Chem. 2000, 275, 23161–23168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, L.B.; Bruun, G.H.; Yang, W.; Treviño, L.R.; Vattathil, S.; Scheet, P.; Cheng, C.; Rosner, G.L.; Giacomini, K.M.; Fan, Y.; et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012, 22, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, C.S.; Bruun, G.H.; Peer, C.J.; Mikkelsen, T.S.; Corydon, T.J.; Gibson, A.A.; Hu, S.; Orwick, S.J.; Mathijssen, R.H.; Figg, W.D.; et al. OATP1B1 polymorphism as a determinant of erythromycin disposition. Clin. Pharmacol. Ther. 2012, 92, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Selen, A.; Amidon, G.L.; Welling, P.G. Pharmacokinetics of probenecid following oral doses to human volunteers. J. Pharm. Sci. 1982, 71, 1238–1242. [Google Scholar] [CrossRef]
- Vasilyeva, A.; Durmus, S.; Li, L.; Wagenaar, E.; Hu, S.; Gibson, A.A.; Panetta, J.C.; Mani, S.; Sparreboom, A.; Baker, S.D.; et al. Hepatocellular Shuttling and Recirculation of Sorafenib-Glucuronide Is Dependent on Abcc2, Abcc3, and Oatp1a/1b. Cancer Res. 2015, 75, 2729–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Panetta, J.C.; Pounds, S.B.; Wang, L.; Li, L.; Navid, F.; Federico, S.M.; Eisenmann, E.D.; Vasilyeva, A.; Wang, Y.; et al. Sorafenib Population Pharmacokinetics and Skin Toxicities in Children and Adolescents with Refractory/Relapsed Leukemia or Solid Tumor Malignancies. Clin. Cancer Res. 2019, 25, 7320–7330. [Google Scholar] [CrossRef] [Green Version]
- Brose, M.S.; Frenette, C.T.; Keefe, S.M.; Stein, S.M. Management of sorafenib-related adverse events: A clinician’s perspective. Semin. Oncol. 2014, 41, S1–S16. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Characteristic | Evaluable Patients (n = 16) |
|---|---|
| Sex | |
| Male | 14 (88%) |
| Female | 2 (12%) |
| Age (years) median [IQR] | 66.5 (58–75) |
| Performance | |
| ECOG 0 | 1 (6%) |
| ECOG 1 | 13 (82%) |
| ECOG 2 | 2 (12%) |
| Tumor type | |
| HCC | 12 (72%) |
| • Liver cirrhosis | 8 (66%) |
| • Pre-existent hepatitis | 5 (42%) |
| Thyroid carcinoma | 4 (28%) |
| BMI (kg/m2) median [IQR] | 25.2 (22–30) |
| Race | |
| Caucasian | 11 (70%) |
| African | 1 (6%) |
| Arabic | 3 (18%) |
| Asian | 1 (6%) |
| Sorafenib daily dose at start of study | |
| 200 mg | 1 (6%) |
| 400 mg | 10 (63%) |
| 600 mg | 2 (12%) |
| 800 mg | 3 (19%) |
| Pharmacokinetic Parameters | Sorafenib Monotherapy | Metabolic Ratio (Metabolite/Sorafenib) | Sorafenib with Probenecid | Metabolic Ratio (Metabolite/Sorafenib) | RD Sorafenib Monotherapy vs. Sorafenib + Probenecid (90% CI) | |
|---|---|---|---|---|---|---|
| Sorafenib | ||||||
| AUC0–12 h (CV%) | 33,457.8 (42) | 24,476.8 (57) | −26.8% (−37.7% to −14.1%) | |||
| geomean μg·h/mL | ||||||
| Cmax (CV%) | 4457.8 (52) | 3337.2 (63) | −25.1% (−44.3% to −19.7%) | |||
| geomean μg/mL | ||||||
| Ctrough (CV%) | 2125.5 (60) | 1571.9 (61) | −26.0% (−43.4% to −3.4%) | |||
| geomean μg/mL | ||||||
| Tmax (IQR) | 3.7 (1.5–4.15) | 2.2 (1.0–2.01) | NA | |||
| median hours | ||||||
| Sorafenib-N-oxide | ||||||
| AUC0–12 h (CV%) | 3442.8 (78) | 0.10 | 2192.3 (77) | 0.09 | −36.3% (−52.8% to −14.1%) | |
| geomean μg·h/mL | ||||||
| Cmax (CV%) | 467.3 (77) | 283.9 (74) | −39.2% (−54.6% to −18.7%) | |||
| geomean μg/mL | ||||||
| Ctrough (CV%) | 271.1 (282) | (71) | −35.2% (−59.7% to 4.3%) | |||
| geomean μg/mL | ||||||
| Sorafenib-glucuronide | ||||||
| AUC0–12 h (CV%) | 120,660 (55) | 3.61 | 113,995 (59) | 4.66 | −5.5% (−18.0% to 8.9%) | |
| geomean μg·h/mL | ||||||
| Cmax (CV%) | 12,704 (51) | 11,931 (64) | −6.1% (−21.7% to 12.7%) | |||
| geomean μg/mL | ||||||
| Ctrough (CV%) | 9159 (65) | 8400 (67) | −8.3% (−21.3% to 6.9%) | |||
| geomean μg/mL | ||||||
| Tissue | ||||||
| Sorafenib concentration in keratinocytes | 50.0 (61) | 1.49 × 10−3 # | 36.0 (63) | 1.47 × 10−3 | −28.1% (−46.3% to −3.7%) | |
| Geomean ng/mL (CV%) | ||||||
| Sorafenib Mono (N = 16) | Sorafenib Concomitantly with Probenecid (N = 16) | |||
|---|---|---|---|---|
| Adverse event | Grade 1–2 | Grade 3 | Grade 1–2 | Grade 3 |
| HFSR | 3 | - | 6 | 1 |
| Rash | 1 | - | 3 | 1 |
| Nausea | 1 | - | 2 | - |
| Vomiting | 0 | - | 1 | - |
| Oral mucositis | 1 | - | 1 | - |
| Diarrhea | 1 | - | 2 | - |
| Constipation | 2 | - | 3 | - |
| Anorexia | 4 | - | 7 | - |
| Dyspnea | - | - | - | 1 |
| Edema | - | - | 1 | - |
| Fatigue | 2 | - | 6 | 1 |
| Fever | 1 | - | - | - |
| Pain | 1 | 1 | 2 | 1 |
| Serious adverse events (SAE) | 1 | 2 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussaarts, K.G.A.M.; van Doorn, L.; Eechoute, K.; Damman, J.; Fu, Q.; van Doorn, N.; Eisenmann, E.D.; Gibson, A.A.; Oomen-de Hoop, E.; de Bruijn, P.; et al. Influence of Probenecid on the Pharmacokinetics and Pharmacodynamics of Sorafenib. Pharmaceutics 2020, 12, 788. https://doi.org/10.3390/pharmaceutics12090788
Hussaarts KGAM, van Doorn L, Eechoute K, Damman J, Fu Q, van Doorn N, Eisenmann ED, Gibson AA, Oomen-de Hoop E, de Bruijn P, et al. Influence of Probenecid on the Pharmacokinetics and Pharmacodynamics of Sorafenib. Pharmaceutics. 2020; 12(9):788. https://doi.org/10.3390/pharmaceutics12090788
Chicago/Turabian StyleHussaarts, Koen G. A. M., Leni van Doorn, Karel Eechoute, Jeffrey Damman, Qiang Fu, Nadia van Doorn, Eric D. Eisenmann, Alice A. Gibson, Esther Oomen-de Hoop, Peter de Bruijn, and et al. 2020. "Influence of Probenecid on the Pharmacokinetics and Pharmacodynamics of Sorafenib" Pharmaceutics 12, no. 9: 788. https://doi.org/10.3390/pharmaceutics12090788
APA StyleHussaarts, K. G. A. M., van Doorn, L., Eechoute, K., Damman, J., Fu, Q., van Doorn, N., Eisenmann, E. D., Gibson, A. A., Oomen-de Hoop, E., de Bruijn, P., Baker, S. D., Koolen, S. L. W., van Gelder, T., van Leeuwen, R. W. F., Mathijssen, R. H. J., Sparreboom, A., & Bins, S. (2020). Influence of Probenecid on the Pharmacokinetics and Pharmacodynamics of Sorafenib. Pharmaceutics, 12(9), 788. https://doi.org/10.3390/pharmaceutics12090788