The Biosimilar Landscape: An Overview of Regulatory Approvals by the EMA and FDA



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Mining
2.1.1. Landscape of EMA and FDA Biosimilar Approvals
2.1.2. Adalimumab Biosimilars Case Study
2.2. Data Analysis and Study Limitations.
3. Results
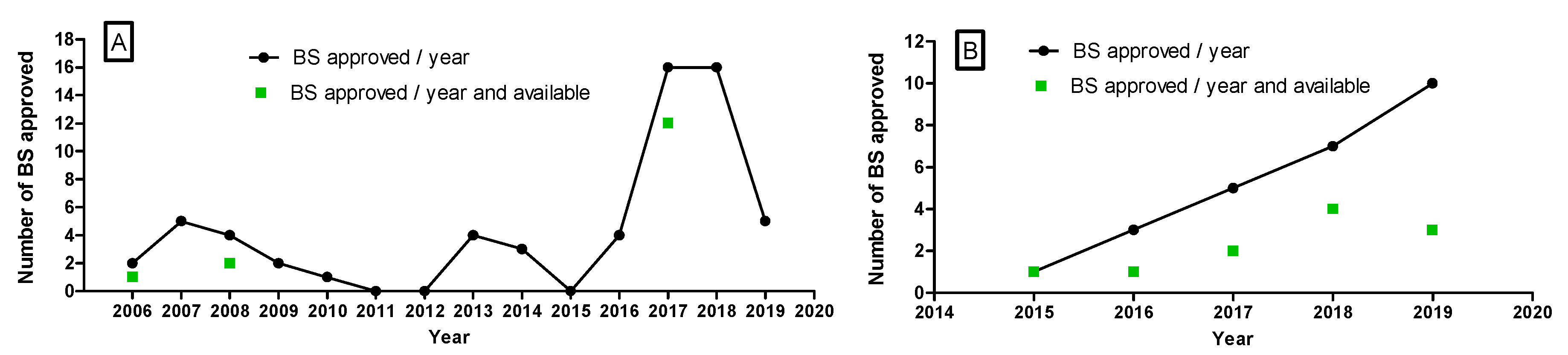
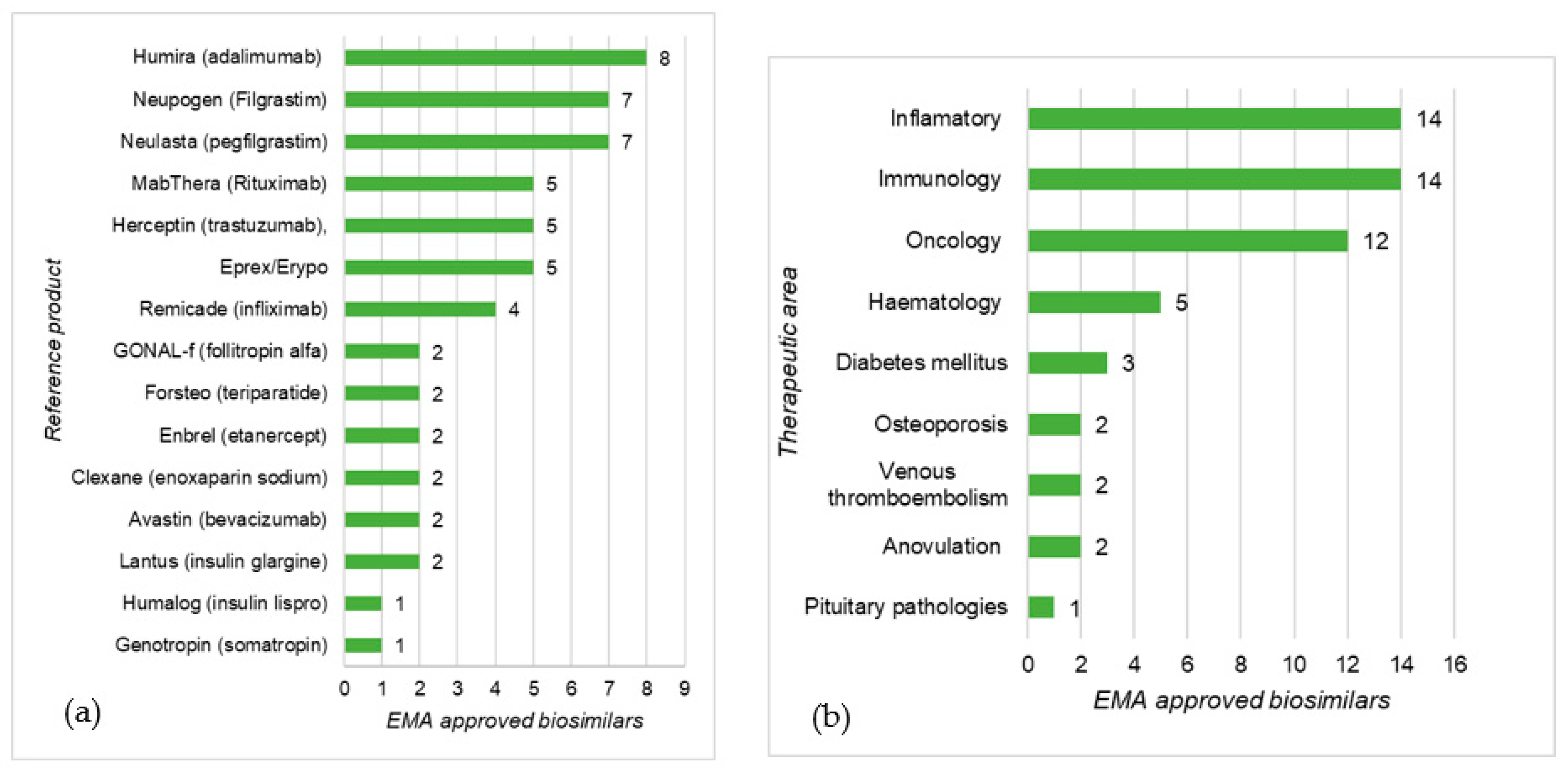
3.1. EMA and FDA Biosimilar Approvals
3.2. Clinical Evidence in EPARs of Adalimumab Biosimilars
4. Discussion
4.1. EMA and FDA Biosimilar Approvals
4.2. Clinical Evidence Package Published in EPARs for Adalimumab Biosimilars
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mehr, S.; Brook, R. Biosimilars in the USA: Will new efforts to spur approvals and access spur uptake and cost savings? Pharm. Med. 2019, 33, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Troein, P.; Newton, M.; Pate, J.; Scott, K. The Impact of Biosimilar Competition in Europe. European Commission. IQVIA Report. October 2019. Available online: https://ec.europa.eu/docsroom/ (accessed on 3 December 2020).
- WHO Questions and Answers: Similar Biotherapeutic Products. Available online: https://www.who.int/biologicals/QA_for_SBPs_HK_12_Dec_2017_(2).pdf22 (accessed on 22 January 2020).
- Daller, J. Biosimilars: A consideration of the regulations in the United States and European Union. Regul. Toxicol. Pharm. 2016, 76, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chow, S. On the regulatory approval pathway of biosimilar products. Pharmaceuticals 2012, 5, 353–368. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. List of Approved NDAs for Biological Products That Were Deemed to Be BLAs on 23 March 2020. Available online: https://www.fda.gov/media/119229/download (accessed on 20 December 2020).
- U.S. Food and Drug Administration. Purple Book Database of Licensed Biological Products. Available online: https://purplebooksearch.fda.gov/ (accessed on 20 December 2020).
- Guideline on Similar Biological Medicinal Products. CHMP/437/04 Rev 1. EMA. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf (accessed on 3 November 2020).
- Biosimilars Action Plan: Balancing Innovation and Competition. Available online: https://www.fda.gov/media/114574/download (accessed on 22 January 2020).
- Multidisciplinary: Biosimilar—EMA. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/multidisciplinary/multidisciplinary-biosimilar (accessed on 22 January 2020).
- World Health Organization. Similar Biotherapeutic Products. Available online: https://www.who.int/biologicals/biotherapeutics/similar_biotherapeutic_products/en/ (accessed on 22 January 2020).
- Bellinvia, S.; Fraser Cummings, J.R.; Ardern-Jones, M.R.; Edwards, C.J. Adalimumab biosimilars in Europe: An overview of the clinical evidence. BioDrugs 2019, 33, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.J.; Mouslim, M.C.; Blunt, J.L.; Alexander, G.C.; Shermock, K.M. Assessment of availability, clinical testing, and US Food and Drug Administration review of biosimilar biologic products. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schiestl, M.; Ranganna, G.; Watson, K.; Jung, B.; Roth, K.; Capsius, B.; Trieb, M.; Bias, P.; Maréchal-Jamil, J. The path towards a tailored clinical biosimilar development. BioDrugs 2020, 34, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff-Holz, E.; Tiitso, K.; Vleminck, C.; Weise, M. Evolution of the EU biosimilar framework: Past and future. BioDrugs 2019, 33, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medicines. European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine/field_ema_med_status/authorised-36/ema_medicine_types/field_ema_med_biosimilar/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar (accessed on 1 November 2019).
- European Commission. Handling of Duplicate Marketing Authorization Applications. Health and Consumers Directorate-General. Available online: https://ec.europa.eu/health/sites/health/files/files/latest_news/2011_09_duplicates_note_upd_01.pdf (accessed on 2 December 2020).
- U.S. Food and Drug Administration. Biosimilar Drug Information. Available online: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information (accessed on 1 November 2019).
- Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 1 November 2019).
- Biosimilars Approved in the US and Filed for FDA Approval. Available online: https://biosimilarsrr.com/us-biosimilar-filings/ (accessed on 4 January 2020).
- Rathore, A.S.; Vulto, A.G.; Stevenson, J.G.; Shah, V.S. Challenges with successful commercialization of biosimilars. BioPharm Int. 2019, 32, 22–31. [Google Scholar]
- Rothwell Figg’s Biologics and Biosimilars Group, Rothwell Figg’s Biosimilars Law Bulletin. How the U.S. Compares to Europe on Biosimilar Approvals and Products in the Pipeline. Available online: https://www.biosimilarsip.com/2020/10/12/how-the-u-s-compares-to-europe-on-biosimilar-approvals-and-products-in-the-pipeline-5/ (accessed on 3 December 2020).
- Sarzi-Puttinia, P.; Marotto, D.; Caporalic, R.; Galeazzid, M.; Atzenie, F.; Hamarf, A.; Soósf, B.; Szekanecz, Z. Biosimilars vs. originators: Are they the same? Autoimmun. Rev. 2019, 18, 102404. [Google Scholar] [CrossRef] [PubMed]
- Enoxaparin Biosimilar or Not. Generics and Biosimilars Initiative. Available online: http://www.gabionline.net/Biosimilars/General/Enoxaparin-biosimilar-or-not#:~:text=A%20biosimilar%20of%20the%20low%20molecular%20weight%20heparin%2C,whether%20this%20was%20really%20a%20biosimilar%20or%20not (accessed on 20 December 2020).
- Guideline on Similar Biological Medicinal Products Containing Monoclonal Antibodies—Non-Clinical and Clinical Issues. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf (accessed on 6 January 2020).
- Age Kos, I.; Azevedo, V.; Neto, D.; Kowalski, S. The biosimilars journey: Current status and ongoing challenges. Drugs Context 2018, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.H.; Yiu, Z.Z.N.; Bale, T.; Burden, A.D.; Coates, L.C.; Edwards, W.; MacMahon, E.; Mahil, S.; McGuire, A.; Murphy, R.; et al. British Association of Dermatologists guidelines for biologic therapy for psoriasis 2020: A rapid update. Br. J. Dermatol. 2020, 183, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Kromeya. CHMP Assessment Report. Available online: https://www.ema.europa.eu/en/documents/assessment-report/kromeya-epar-public-assessment-report_en.pdf (accessed on 23 January 2020).
- Pelechas, E.; Drosos, A. Etanercept biosimilar SB-4. Expert Opin. Biol. Ther. 2019, 19, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Barbier, L.; Declerck, P.; Simoens, S.; Neven, P.; Vulto, A.; Huys, I. The arrival of biosimilar monoclonal antibodies in oncology: Clinical studies for trastuzumab biosimilars. Br. J. Cancer 2019, 121, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speights, K. Biggest Blockbuster Drugs of the Future. The Motley Fool. Available online: https://www.fool.com/investing/2019/06/15/5-biggest-blockbuster-drugs-of-the-future.aspx (accessed on 23 January 2020).
- Amgen’s Adalimumab Biosimilar Will Only Be Launched in US in 2023. Generics and Biosimilars Initiative. Available online: http://gabionline.net/Biosimilars/News/Amgen-s-adalimumab-biosimilar-will-only-be-launched-in-US-in-2023#:~:text=The%20settlement%20means%20that%20Amgen%E2%80%99s%20adalimumab%20biosimilar%20will,launched%20in%20the%20US%20on%2031%20January%202023 (accessed on 20 December 2020).
- Sandoz Announces Global Resolution of Biosimilar Adalimumab Patent Disputes, Securing Patient Access. Available online: https://www.sandoz.com/news/media-releases/sandoz-announces-global-resolution-biosimilar-adalimumab-patent-disputes (accessed on 23 January 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Product | Biosimilar Name | Approval Date | Biosimilar Name | Approval Date | |
|---|---|---|---|---|---|
| MabThera (rituximab) | Truxima (rituximab) | 17 February 2017 | Rixathon (rituximab) | 15 June 2017 | |
| Ritemvia (rituximab) | 13 July 2017 | Riximyo (rituximab) | 15 June 2017 | ||
| Blitzima (rituximab) | 13 July 2017 | ||||
| Eprex/Erypo (epoetin alfa) | Epoetin Alfa Hexal | 27 August 2007 | |||
| Binocrit (epoetin alfa) | 28 August 2007 | ||||
| Abseamed (epoetin alfa) | 27 August 2007 | ||||
| Eprex/Erypo (epoetin zeta) | Retacrit (epoetin zeta) | 18 December 2007 | |||
| Silapo (epoetin zeta) | 18 December 2007 | ||||
| Herceptin (trastuzumab) | Ontruzant (trastuzumab) | 15 November 2017 | Kanjinti (trastuzumab) | 16 May 2018 | |
| Trazimera (trastuzumab) | 26 July 2018 | Ogivri (trastuzumab) | 12 December 2018 | ||
| Herzuma (trastuzumab) | 8 February 2018 | ||||
| Neupogen (filgrastim) | Nivestim (filgrastim) | 7 June 2010 | Grastofil (filgrastim) | 17 October 2013 | |
| Filgrastim Hexal (filgrastim) | 6 February 2009 | Tevagrastim (filgrastim) | 15 September 2008 | ||
| Zarzio (filgrastim) | 6 February 2009 | Ratiograstim (filgrastim) | 15 September 2008 | ||
| Accofil (filgrastim) | 17 September 2014 | ||||
| Neulasta (pegfilgrastim) | Pelmeg (pegfilgrastim) | 20 November 2018 | Ziextenzo (pegfilgrastim) | 22 November 2018 | |
| Udenyca (pegfilgrastim) | 21 September 2018 | Grasustek (pegfilgrastim) | 20 June 2019 | ||
| Fulphila (pegfilgrastim) | 20 November 2018 | Cegfila (pegfilgrastim) | 19 December 2019 | ||
| Pelgraz (pegfilgrastim) | 21 September 2018 | ||||
| Remicade (infliximab) | Remsima (infliximab) | 10 September 2013 | Zessly (infliximab) | 18 May 2018 | |
| Inflectra (infliximab) | 9 September 2013 | ||||
| Flixabi (infliximab) | 26 May 2016 | ||||
| Enbrel (etanercept) | Benepali (etanercept) | 13 January 2016 | |||
| Erelzi (etanercept) | 23 June 2017 | ||||
| GONAL-f (follitropin alfa) | Ovaleap (follitropin alfa) | 27 September 2013 | |||
| Bemfola (follitropin alfa) | 26 March 2014 | ||||
| Avastin (bevacizumab) | Zirabev (bevacizumab) | 14 February 2019 | |||
| Mvasi (bevacizumab) | 15 January 2018 | ||||
| Humira (adalimumab) | Hefiya (adalimumab) | 26 July 2018 | Amgevita (adalimumab) | 21 March 2017 | |
| Kromeya (adalimumab) | 2 April 2019 | Idacio (adalimumab) | 2 April 2019 | ||
| Imraldi (adalimumab) | 24 August 2017 | Halimatoz (adalimumab) | 26 July 2018 | ||
| Hyrimoz (adalimumab) | 26 July 2018 | Hulio (adalimumab) | 16 September 2018 | ||
| Lantus (insulin glargine) | Semglee (insulin glargine) | 23 March 2018 | |||
| Abasaglar (insulin glargine) | 09 September 2014 | ||||
| Humalog (insulin lispro) | Insulin lispro Sanofi (insulin lispro) | 18 July 2017 | |||
| Genotropin (somatropin) | Omnitrope (somatropin) | 12 April 2006 | |||
| Clexane (enoxaparin sodium) | Thorinane (enoxaparin sodium) | 14 September 2016 | |||
| Inhixa (enoxaparin sodium) | 15 September 2016 | ||||
| Forsteo (teriparatide) | Terrosa (teriparatide) | 4 January 2017 | |||
| Movymia (teriparatide) | 11 January 2017 | ||||
| Reference Product | Biosimilar Name | Approval Date | Market Status |
|---|---|---|---|
| Avastin (bevacizumab) | Mvasi (bevacizumab-awwb) | 14 September 2017 | Available |
| Zirabev (bevacizumab-bvzr) | 27 June 2019 | Available | |
| Enbrel (etanercept) | Erelzi (etanercept-szzs) | 30 August 2016 | Not available |
| Eticovo (etanercept-ykro) | 25 April 2019 | Not available | |
| Epogen (epoetin-alfa) | Retacrit (epoetin alfa-epbx) | 15 May 2018 | Available |
| Herceptin (trastuzumab) | Ogivri (trastuzumab-dkst) | 1 December 2017 | Not available |
| Herzuma (trastuzumab-pkrb) | 14 December 2018 | Not available | |
| Ontruzant (trastuzumab-dttb) | 18 January 2019 | Not available | |
| Trazimera (trastuzumab-qyyp) | 11 March 2019 | Not available | |
| Kanjinti (trastuzumab-anns) | 13 June 2019 | Available | |
| Humira (adalimumab) | Amjevita (adalimumab-atto) | 23 September 2016 | Not available |
| Cyltezo (adalimumab-adbm) | 25 August 2017 | Not available | |
| Hyrimoz (adalimumab-adaz) | 30 October 2018 | Not available | |
| Hadlima (adalimumab-bwwd) | 23 July 2019 | Not available | |
| Abrilada (adalimumab-afzb) | 15 November 2019 | Not available | |
| Neulasta (pegfilgrastim) | Fulphila (pegfilgrastim-jmdb) | 4 June 2018 | Available |
| Udenyca (pegfilgrastim-cbqv) | 2 November 2018 | Available | |
| Ziextenzo (pegfilgastrim-bmez) | 4 November 2019 | Available | |
| Neupogen (filgrastim) | Zarxio (filgrastim-sndz) | 6 March 2015 | Available |
| Nivestym (filgrastim-aafi) | 20 July 2018 | Available | |
| Remicade (infliximab) | Inflectra (infliximab-dyyb) | 5 April 2016 | Available |
| Ixifi (infliximab-qbtx) | 13 December 2017 | Not available | |
| Renflexis (infliximab-abda) | 21 April 2017 | Available | |
| Avsola (infliximab-axxq) | 6 December 2019 | Not available | |
| Rituxan (rituximab) | Truxima (rituximab-abbs) | 28 November 2018 | Not available |
| Ruxience (rituximab-pvvr) | 23 July 2019 | Not available |
| Company | Molecule | Brand Name | Approval Date | Phase I Studies (N) | Phase III Studies | ||
|---|---|---|---|---|---|---|---|
| EU | FDA | (N) | Investigated Conditions | ||||
| Sandoz | GP2017 | EU Hyrimoz (adalimumab) | July 2018 | NA | 3 | 1 | Plaque psoriasis |
| US Hyrimoz (adalimumab-adaz) | NA | October 2018 | |||||
| Halimatoz (adalimumab) | July 2018 | NA | |||||
| Hefiya (adalimumab) | July 2018 | NA | |||||
| Amgen | ABP 501 | Amgevita (adalimumab) | March 2017 | NA | 1 | 2 | RA and psoriasis |
| Amjevita (adalimumab-atto) | NA | September 2016 | |||||
| Samsung Bioepis NL B.V. | SB5 | Imraldi (adalimumab) | August 2017 | NA | 1 | 1 | RA |
| Hadlima (adalimumab-bwwd) | NA | July 2019 | |||||
| Fresenius Kabi Deutschland GmbH | MSB11022 | Kromeya (adalimumab) | April 2019 | NA | 1 | 1 | Plaque psoriasis |
| Idacio (adalimumab) | April 2019 | NA | |||||
| Mylan S.A.S. | FKB327 | Hulio (adalimumab) | September 2018 | NA | 2 | 1 | RA |
| Company | Molecule | Study Number | Duration (Days) | N * | Comparing | Comparer | Primary PK Endpoints |
|---|---|---|---|---|---|---|---|
| Amgen | ABP 501 | 20110217 | 63 | 203 | PK profile | Adalimumab EU and US | AUC0–inf, AUC0–t, Cmax |
| Sandoz | GP2017 | GP17-104 | 72 | 318 | PK profile | Adalimumab EU and US | AUC0–inf, Cmax |
| GP17-101 | 72 | 219 | PK profile | Adalimumab EU and US | AUC0–inf, AUC0–last, Cmax | ||
| GP17-102 | 72 | 108 | PK profile | AI vs. PFS administration | AUC0–360 h, Cmax | ||
| Samsung Bioepis NL B.V. | SB5 | SB5-G11-NHV | 70 | 189 | PK profile | Adalimumab EU and US | EMA: AUCinf, Cmax FDA: AUCinf, AUClast, Cmax |
| Fresenius Kabi Deutschland GmbH | MSB11022 | EMR200588-001 | 70 | 233 | PK profile | Adalimumab EU and US | AUC0–inf, AUC0–t, Cmax |
| Mylan S.A.S. | FKB327 | FKB327-001 | 64 | 180 | PK profile | Adalimumab EU and US | AUC0–last, Cmax |
| FKB327-005 | 64 | 129 | Relative bioavailability | Delivery via vial, PFS and AI | AUC0–t, AUC0–inf, Cmax |
| Molecule | Study Number | Studied Population | N * | Comparison | Primary Efficcay Endpoint | Equivalence Margin | Primary Endpoint Results | 95% CI |
|---|---|---|---|---|---|---|---|---|
| ABP 501 | 20120262 | Adults with moderate to severe RA | 526 | ABP 501 vs. adalimumab (US) | Risk ratio of ACR20 at week 24 | [0.738; 1/0.738] | ACR20 response rate 74.6% vs. 72.4%, risk ratio of ACR20 ABP501 vs. Humira 1.039 | (0.954, 1.133) |
| 20120263 | Adults with moderate to severe psoriasis | 350 | ABP 501 with adalimumab (EU) | PASI% improvement from baseline at week 16 | [−15%; 15%] | PASI% improvement 80.91%(ABP 501) vs. 83.06% (Humira)–difference in response −2.18 | (−7.39, 3.02) | |
| GP2017 | GP17-301 | Male and female patients with moderate to severe chronic plaque-type psoriasis | 465 | GP2017 vs. EU-Humira and US-Humira | PASI75 response rate at week 16 | [−18%; 18%] | 66.8% for GP2017 and 65.0% for Humira | (−7.46, 11.15) |
| SB5 | SB5-G31-RA | Adults with moderate to severe rheumatoid arthritis despite methrotexate | 544 | SB5 vs. EU Humira | ACR20 response rate at week 24 | [−15%; 15%] | ACR20 response rate at week 24 68.0% (183/269) SB5 and 67.4% (184/273) Humira | (−7.83, 8.13) |
| MSB11022 | EMR200588-022 | Patients with moderate to severe chronic plaque psoriasis | 382 | MSB11022 vs. EU-approved Humira | PASI75 at week 16 | [−18%; 18%] | PASI75 response at week 16: 90% in MSB1102 group and 92% in EU-Humira group | (−7.82, 4.16) |
| FKB327 | FKB327-022 followed by FKB327-003 | Rheumatoid arthritis patients inadequately controlled on methotrexate | 680 | FKB327 vs. US-Humira | ACR20 response rate at week 24 | [−13%; 13%] | 270 patients (74.4%) in the FKB327 treatment group achieved an ACR20 response at week 24 compared to 271 patients (75.7%) | (−7.6, 5.0) |
| Pharmaceutical Company | ABBVIE | SANDOZ | AMGEN | Samsung Bioepis NL B.V. | Fresenius Kabi Deutchsland | MYLAN | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Indication | EU Humira | US Humira | EU Hyrimoz | US Hyrimoz | Hefiya | Halimatoz | Amgevita | Amjevita | Imraldi | Hadlima | Kromeya | Idacio | Hulio |
| Rheumatoid Arthritis | yes | yes | yes | yes | no | yes | yes | yes | yes | yes | yes | yes | yes |
| Juvenile idiopathic arthitis | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes |
| Ankylosing spondylitis (AS) | yes | yes | yes | yes | yes | yes | yes | no | yes | yes | yes | yes | yes |
| Psoriatic Arthritis | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes |
| Plaque Psoriasis | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes |
| Paediatric Plaque Psoriasis | yes | no | yes | no | yes | yes | yes | no | yes | no | yes | yes | yes |
| Hidradenitis suppurativa (HS) | yes | yes | yes | no | yes | yes | yes | no | yes | no | no | yes | yes |
| Crohn’s Disease | yes | yes | yes | yes | no | no | yes | yes | yes | yes | yes | yes | yes |
| Pediatric Crohn’s Disease | yes | yes | yes | no | no | no | yes | no | yes | no | yes | yes | yes |
| Ulcerative Colitis | yes | yes | yes | yes | no | no | yes | yes | yes | no | yes | yes | yes |
| Uveitis | yes | yes | yes | no | yes | yes | yes | no | yes | no | yes | yes | yes |
| Paediatric uveitis | yes | yes | yes | no | yes | yes | yes | no | yes | no | yes | yes | yes |
| LABEL | Full | Reduced | Full | Reduced | Reduced | Reduced | Full | Reduced | Full | Reduced | Reduced | Full | Full |
| Number of indications | 12 | 11 | 12 | 7 | 8 | 9 | 12 | 6 | 12 | 6 | 11 | 12 | 12 |
| Regulatory jurisdiction | EU | US | EU | US | EU | EU | EU | US | EU | US | EU | EU | EU |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gherghescu, I.; Delgado-Charro, M.B. The Biosimilar Landscape: An Overview of Regulatory Approvals by the EMA and FDA. Pharmaceutics 2021, 13, 48. https://doi.org/10.3390/pharmaceutics13010048
Gherghescu I, Delgado-Charro MB. The Biosimilar Landscape: An Overview of Regulatory Approvals by the EMA and FDA. Pharmaceutics. 2021; 13(1):48. https://doi.org/10.3390/pharmaceutics13010048
Chicago/Turabian StyleGherghescu, Ioana, and M. Begoña Delgado-Charro. 2021. "The Biosimilar Landscape: An Overview of Regulatory Approvals by the EMA and FDA" Pharmaceutics 13, no. 1: 48. https://doi.org/10.3390/pharmaceutics13010048
APA StyleGherghescu, I., & Delgado-Charro, M. B. (2021). The Biosimilar Landscape: An Overview of Regulatory Approvals by the EMA and FDA. Pharmaceutics, 13(1), 48. https://doi.org/10.3390/pharmaceutics13010048