
Enhanced Detection of Desmoplasia by Targeted Delivery of Iron Oxide Nanoparticles to the Tumour-Specific Extracellular Matrix

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
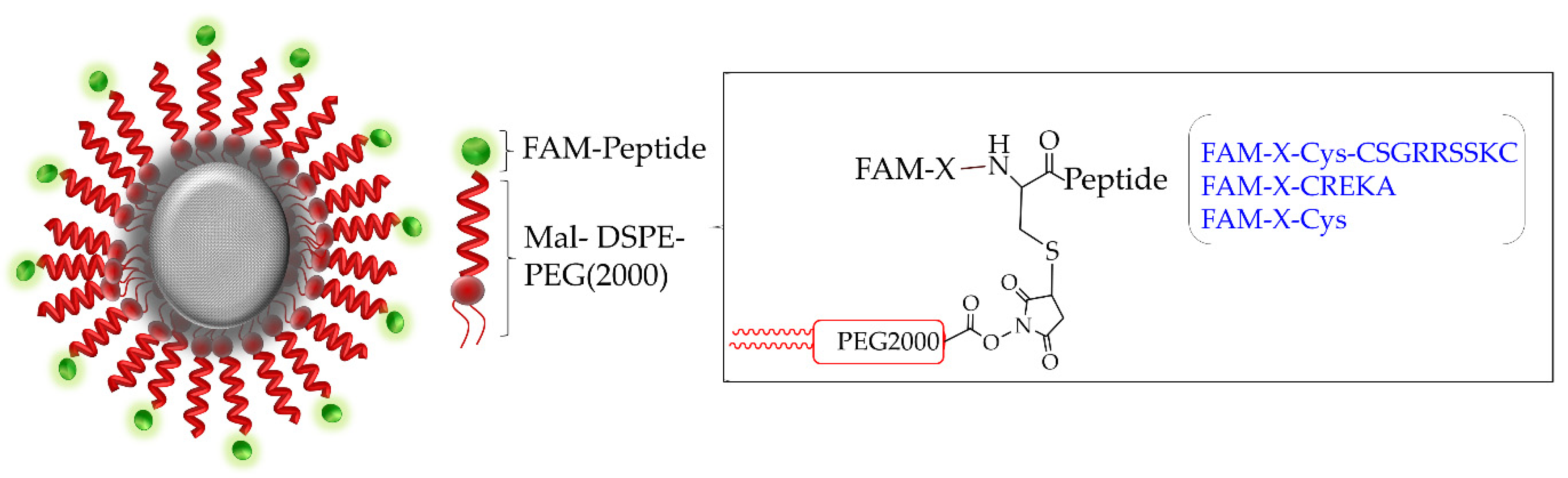
2.1. Peptide
2.2. PNET Mouse Model
2.3. In Vivo Peptide Binding
2.4. Synthesis of Targeted Iron Oxide Nanoparticles
2.5. Material Characterisation
2.6. In Vitro IO-NP MRI
2.7. Ex Vivo Tissue MRI
2.8. In Vivo Lectin Perfusion
2.9. Immunofluorescence and Immunohistochemistry Analysis
2.10. Statistical Analysis
3. Results
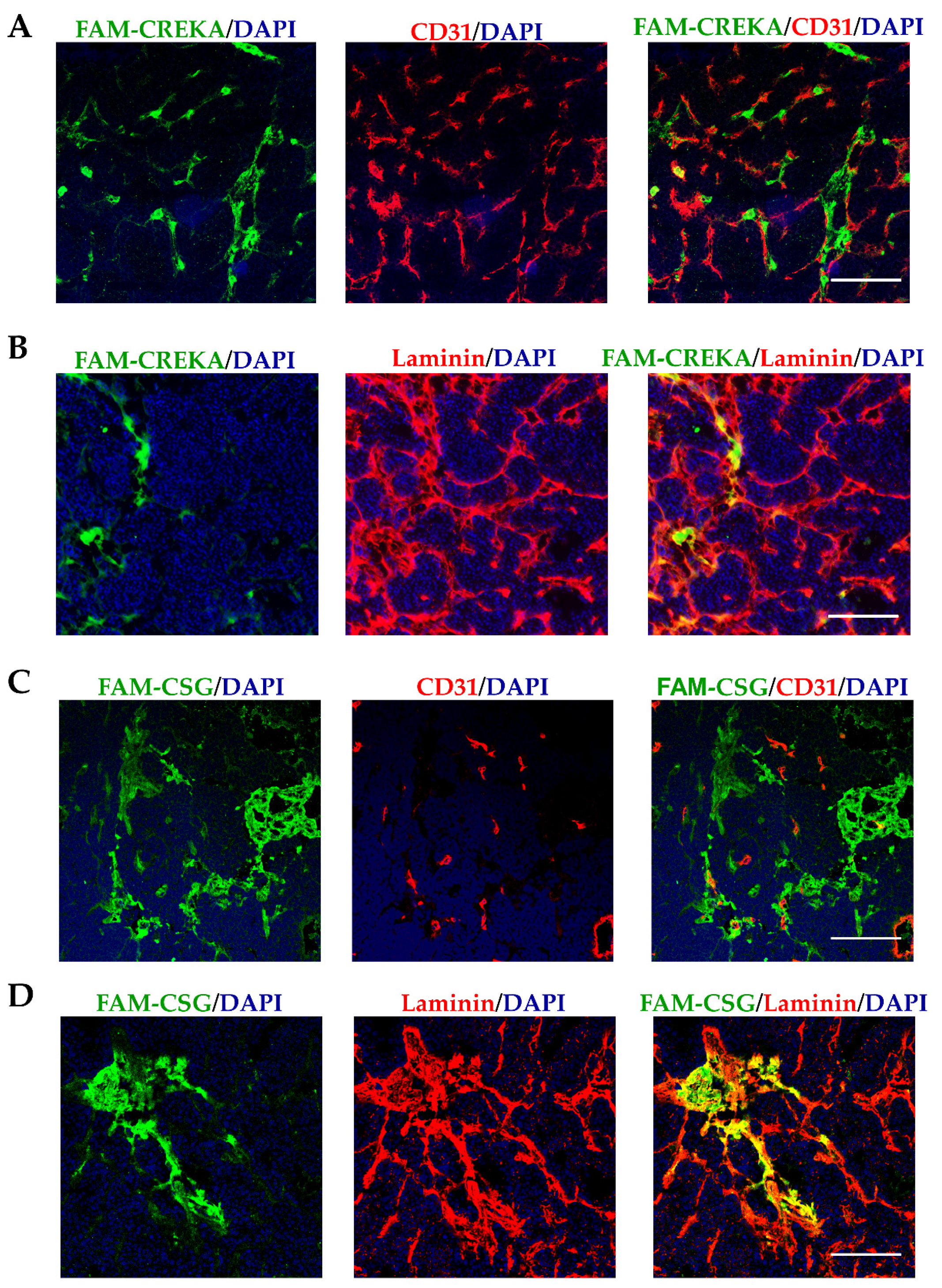
3.1. CSG and CREKA Have Distinct Binding Targets in PNET In Vivo
3.2. Synthesis and Characterization of IO-NP
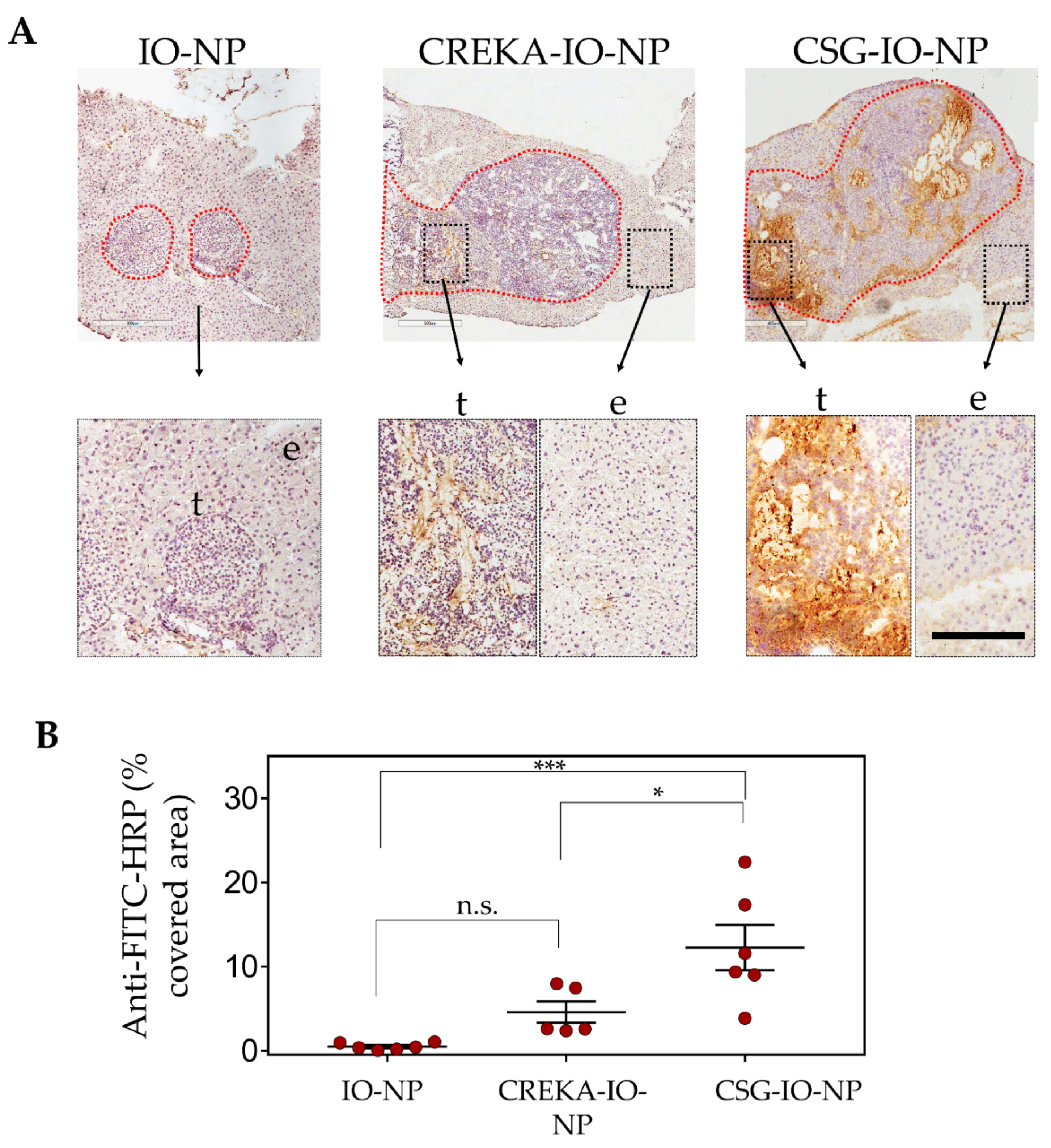
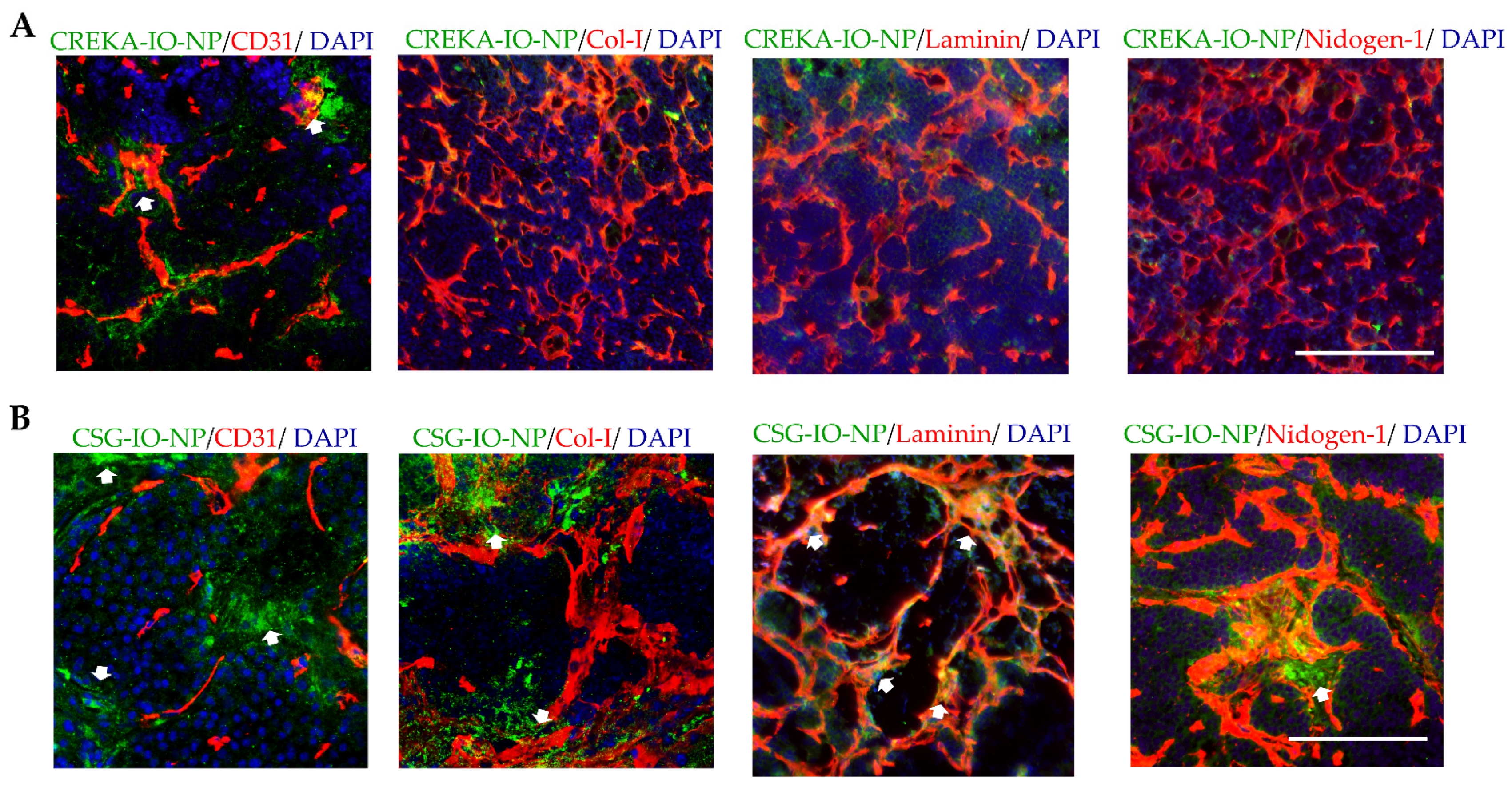
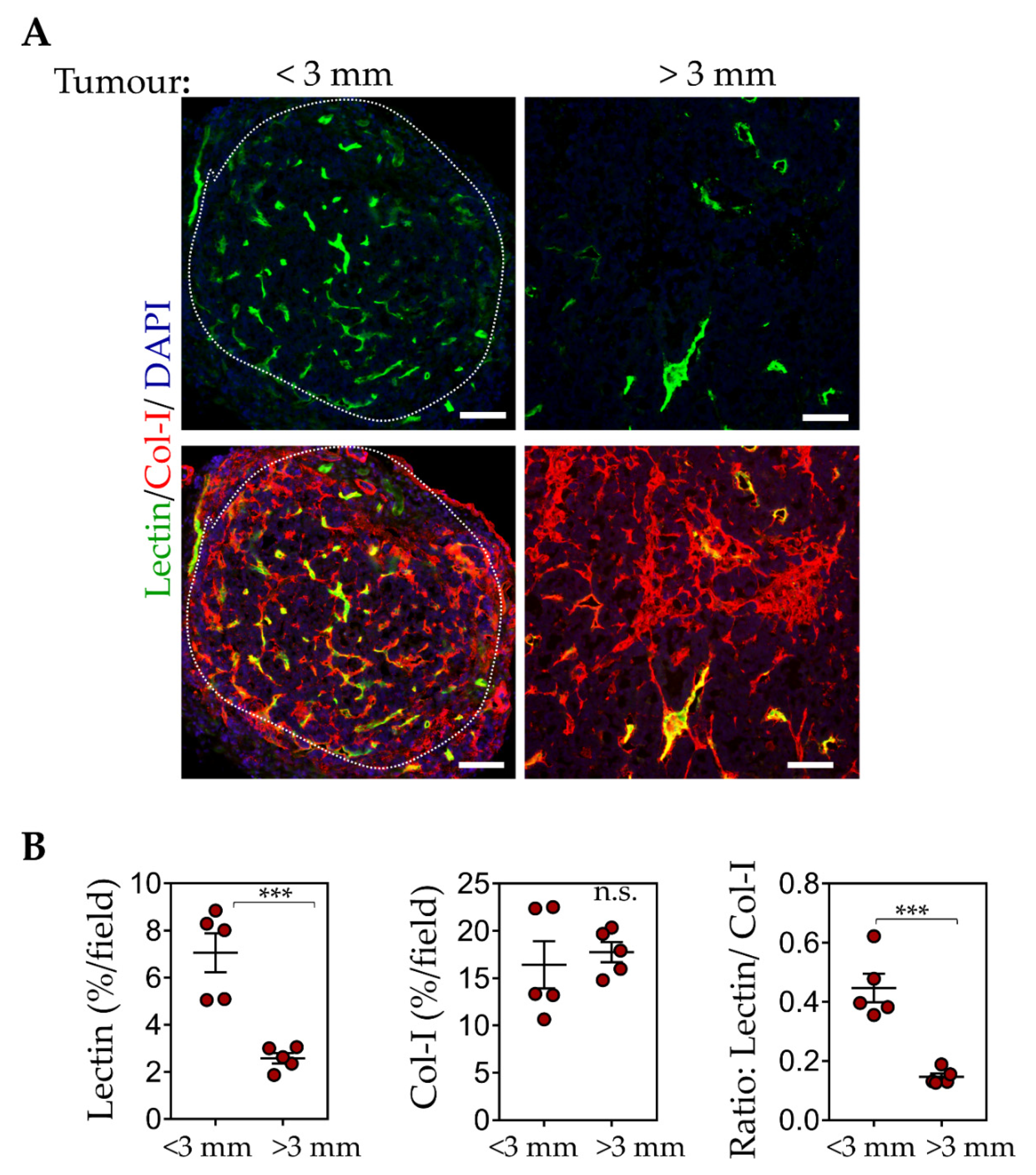
3.3. Enhanced Intratumoral ECM Binding of CSG-IO-NP Compared to Untargeted IO-NP and CREKA-IO-NP
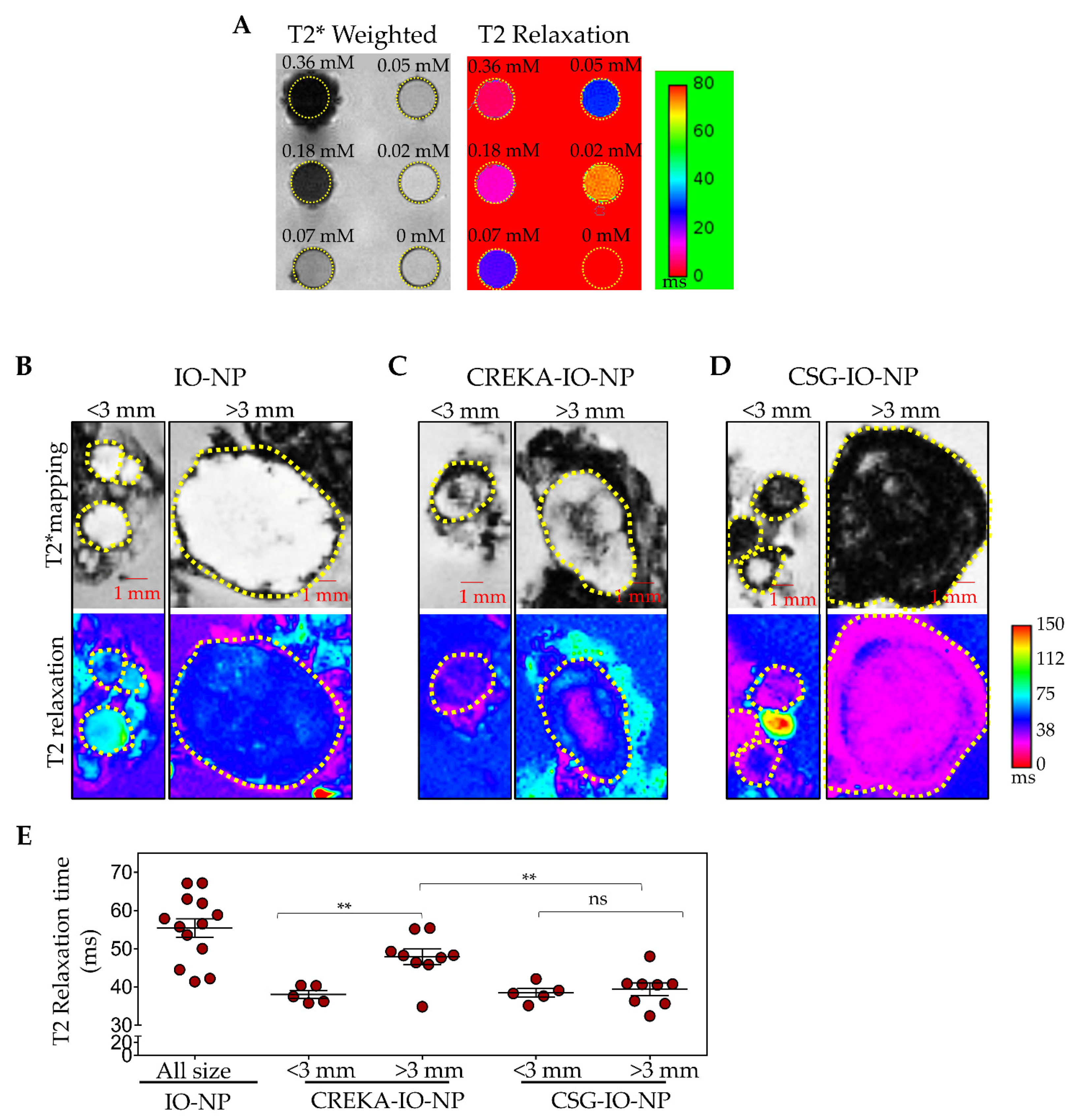
3.4. CSG-IO-NP Is an Effective MRI Contrast Agent, Irrespective of Tumour Sizes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schneble, E.J.; Graham, L.J.; Shupe, M.P.; Flynt, F.L.; Banks, K.P.; Kirkpatrick, A.D.; Nissan, A.; Henry, L.; Stojadinovic, A.; Shumway, N.M.; et al. Current approaches and challenges in early detection of breast cancer recurrence. J. Cancer 2014, 5, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early detection of pancreatic cancer: Opportunities and challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Sanagapalli, S.; Stoita, A. Challenges in diagnosis of pancreatic cancer. World J. Gastroenterol. 2018, 24, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Du, Y.; Ye, J.; Kou, D.; Qiu, J.; Wang, J.; Tian, J.; Chen, X. Intraoperative imaging-guided cancer surgery: From current fluorescence molecular imaging methods to future multi-modality imaging technology. Theranostics 2014, 4, 1072–1084. [Google Scholar] [CrossRef] [Green Version]
- Madajewski, B.; Judy, B.F.; Mouchli, A.; Kapoor, V.; Holt, D.; Wang, M.D.; Nie, S.; Singhal, S. Intraoperative near-infrared imaging of surgical wounds after tumor resections can detect residual disease. Clin. Cancer Res. 2012, 18, 5741–5751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, A.; De, S. Drivers of dynamic intratumor heterogeneity and phenotypic plasticity. Am. J. Physiol. Cell Physiol. 2021, 320, C750–C760. [Google Scholar] [CrossRef] [PubMed]
- Vickman, R.E.; Faget, D.V.; Beachy, P.; Beebe, D.; Bhowmick, N.A.; Cukierman, E.; Deng, W.M.; Granneman, J.G.; Hildesheim, J.; Kalluri, R.; et al. Deconstructing tumor heterogeneity: The stromal perspective. Oncotarget 2020, 11, 3621–3632. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lee, S.J.; Yoon, S.; Ryoo, B.Y.; Kim, S.W.; Choi, S.H.; Lee, S.M.; Chae, E.J.; Park, Y.; Jang, S.J.; et al. Feasibility, safety, and adequacy of research biopsies for cancer clinical trials at an academic medical center. PLoS ONE 2019, 14, e0221065. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.X. Superparamagnetic iron oxide based MRI contrast agents: Current status of clinical application. Quant. Imaging Med. Surg. 2011, 1, 35–40. [Google Scholar]
- Ruoslahti, E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv. Mater. 2012, 24, 3747–3756. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Targeting of drugs and nanoparticles to tumors. J. Cell Biol. 2010, 188, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasqualini, R.; Koivunen, E.; Ruoslahti, E. Alpha v integrins as receptors for tumor targeting by circulating ligands. Nat. Biotechnol. 1997, 15, 542–546. [Google Scholar] [CrossRef]
- Ruoslahti, E. The RGD story: A personal account. Matrix Biol. 2003, 22, 459–465. [Google Scholar] [CrossRef]
- Joyce, J.A.; Laakkonen, P.; Bernasconi, M.; Bergers, G.; Ruoslahti, E.; Hanahan, D. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 2003, 4, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 8, 751–755. [Google Scholar] [CrossRef]
- Laakkonen, P.; Zhang, L.; Ruoslahti, E. Peptide targeting of tumor lymph vessels. Ann. N. Y. Acad. Sci. 2008, 1131, 37–43. [Google Scholar] [CrossRef]
- Pasqualini, R.; Koivunen, E.; Kain, R.; Lahdenranta, J.; Sakamoto, M.; Stryhn, A.; Ashmun, R.A.; Shapiro, L.H.; Arap, W.; Ruoslahti, E. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000, 60, 722–727. [Google Scholar]
- Pilch, J.; Brown, D.M.; Komatsu, M.; Jarvinen, T.A.; Yang, M.; Peters, D.; Hoffman, R.M.; Ruoslahti, E. Peptides selected for binding to clotted plasma accumulate in tumor stroma and wounds. Proc. Natl. Acad. Sci. USA 2006, 103, 2800–2804. [Google Scholar] [CrossRef] [Green Version]
- Roth, L.; Agemy, L.; Kotamraju, V.R.; Braun, G.; Teesalu, T.; Sugahara, K.N.; Hamzah, J.; Ruoslahti, E. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene 2012, 31, 3754–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simberg, D.; Duza, T.; Park, J.H.; Essler, M.; Pilch, J.; Zhang, L.; Derfus, A.M.; Yang, M.; Hoffman, R.M.; Bhatia, S.; et al. Biomimetic amplification of nanoparticle homing to tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 932–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemons, T.D.; Singh, R.; Sorolla, A.; Chaudhari, N.; Hubbard, A.; Iyer, K.S. Distinction between active and passive targeting of nanoparticles dictate their overall therapeutic efficacy. Langmuir 2018, 34, 15343–15349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agemy, L.; Friedmann-Morvinski, D.; Kotamraju, V.R.; Roth, L.; Sugahara, K.N.; Girard, O.M.; Mattrey, R.F.; Verma, I.M.; Ruoslahti, E. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17450–17455. [Google Scholar] [CrossRef] [Green Version]
- Agemy, L.; Sugahara, K.N.; Kotamraju, V.R.; Gujraty, K.; Girard, O.M.; Kono, Y.; Mattrey, R.F.; Park, J.H.; Sailor, M.J.; Jimenez, A.I.; et al. Nanoparticle-induced vascular blockade in human prostate cancer. Blood 2010, 116, 2847–2856. [Google Scholar] [CrossRef] [Green Version]
- Hamzah, J.; Altin, J.G.; Herringson, T.; Parish, C.R.; Hammerling, G.J.; O’Donoghue, H.; Ganss, R. Targeted liposomal delivery of TLR9 ligands activates spontaneous antitumor immunity in an autochthonous cancer model. J. Immunol. 2009, 183, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.; Hamzah, J.; Payne, C.J.; Ganss, R. Tumor-targeted TNFalpha stabilizes tumor vessels and enhances active immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 7841–7846. [Google Scholar] [CrossRef] [Green Version]
- Johansson-Percival, A.; Li, Z.J.; Lakhiani, D.D.; He, B.; Wang, X.; Hamzah, J.; Ganss, R. Intratumoral LIGHT restores pericyte contractile properties and vessel integrity. Cell Rep. 2015, 13, 2687–2698. [Google Scholar] [CrossRef] [Green Version]
- Karmali, P.P.; Kotamraju, V.R.; Kastantin, M.; Black, M.; Missirlis, D.; Tirrell, M.; Ruoslahti, E. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomedicine 2009, 5, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; von Maltzahn, G.; Zhang, L.; Derfus, A.M.; Simberg, D.; Harris, T.J.; Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Systematic surface engineering of magnetic nanoworms for in vivo tumor targeting. Small 2009, 5, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 2010, 328, 1031–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, R.; Chakraborty, S.; Dash, A. Molecular imaging of breast cancer: Role of RGD peptides. Mini Rev. Med. Chem. 2015, 15, 1073–1094. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Niu, G.; Wu, H.; Chen, X. Clinical application of radiolabeled RGD peptides for PET imaging of integrin alphavbeta3. Theranostics 2016, 6, 78–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.J.; Li, H.S.; Wang, Q.S.; Wu, H.B.; Han, Y.J.; Zhou, W.L.; Wang, M.; Huang, S. Construction and evaluation of the tumor-targeting, cell-penetrating multifunctional molecular probe iCREKA. Contrast Media Mol. Imaging 2018, 2018, 7929617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.E.; Niu, Y.; Wu, H.; Amin, M.N.; Cai, J. Development of NGR peptide-based agents for tumor imaging. Am. J. Nucl. Med. Mol. Imaging 2011, 1, 36–46. [Google Scholar] [PubMed]
- Zhou, Z.; Qutaish, M.; Han, Z.; Schur, R.M.; Liu, Y.; Wilson, D.L.; Lu, Z.R. MRI detection of breast cancer micrometastases with a fibronectin-targeting contrast agent. Nat. Commun. 2015, 6, 7984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillies, R.J.; Schornack, P.A.; Secomb, T.W.; Raghunand, N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia 1999, 1, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Henke, E.; Nandigama, R.; Ergun, S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front. Mol. Biosci. 2019, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Leppanen, J.; Lindholm, V.; Isohookana, J.; Haapasaari, K.M.; Karihtala, P.; Lehenkari, P.P.; Saarnio, J.; Kauppila, J.H.; Karttunen, T.J.; Helminen, O.; et al. Tenascin C, fibronectin, and tumor-stroma ratio in pancreatic ductal adenocarcinoma. Pancreas 2019, 48, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma involvement in pancreatic ductal adenocarcinoma: An overview focusing on extracellular matrix proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.K.; Strauss, R.; Richter, M.; Yun, C.O.; Lieber, A. Strategies to increase drug penetration in solid tumors. Front. Oncol. 2013, 3, 193. [Google Scholar] [CrossRef] [Green Version]
- Stern, L.A.; Jonsson, V.D.; Priceman, S.J. CAR T cell therapy progress and challenges for solid tumors. Cancer Treat. Res. 2020, 180, 297–326. [Google Scholar]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.L.; Carretta, M.; Kalvisa, A.; Siersbaek, M.S.; Simoes, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeow, Y.L.; Kotamraju, V.R.; Wang, X.; Chopra, M.; Azme, N.; Wu, J.; Schoep, T.D.; Delaney, D.S.; Feindel, K.; Li, J.; et al. Immune-mediated ECM depletion improves tumour perfusion and payload delivery. EMBO Mol. Med. 2019, 11, e10923. [Google Scholar] [CrossRef]
- Ganss, R.; Hanahan, D. Tumor microenvironment can restrict the effectiveness of activated antitumor lymphocytes. Cancer Res. 1998, 58, 4673–4681. [Google Scholar] [PubMed]
- Kang, Y.S.; Risbud, S.; Rabolt, J.F.; Stroeve, P. Synthesis and characterization of nanometer-size Fe3O4 and γ-Fe2O3 particles. Chem. Mater. 1996, 8, 2209–2211. [Google Scholar] [CrossRef]
- Yu, M.; Huang, S.; Yu, K.J.; Clyne, A.M. Dextran and polymer polyethylene glycol (PEG) coating reduce both 5 and 30 nm iron oxide nanoparticle cytotoxicity in 2D and 3D cell culture. Int. J. Mol. Sci. 2012, 13, 5554–5570. [Google Scholar] [CrossRef] [Green Version]
- Starmans, L.W.; Burdinski, D.; Haex, N.P.; Moonen, R.P.; Strijkers, G.J.; Nicolay, K.; Grull, H. Iron oxide nanoparticle-micelles (ION-micelles) for sensitive (molecular) magnetic particle imaging and magnetic resonance imaging. PLoS ONE 2013, 8, e57335. [Google Scholar] [CrossRef] [Green Version]
- Stoyanova, A.M. Spectrophotometric determination of trace iron by using its catalytic effect on the N-phenylanthranilic acid-potassium periodate reaction. Anal. Sci. 2008, 24, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Zeng, H.; Robinson, D.B.; Raoux, S.; Rice, P.M.; Wang, S.X.; Li, G. Monodisperse MFe2O4 (M = Fe, Co, Mn) nanoparticles. J. Am. Chem. Soc. 2004, 126, 273–279. [Google Scholar] [CrossRef]
- Moradi Khaniabadi, P.; Shahbazi-Gahrouei, D.; Jaafar, M.S.; Majid, A.; Moradi Khaniabadi, B.; Shahbazi-Gahrouei, S. Magnetic iron oxide nanoparticles as T2 MR imaging contrast agent for detection of breast cancer (MCF-7) cell. Avicenna J. Med. Biotechnol. 2017, 9, 181–188. [Google Scholar]
- Stephen, Z.R.; Kievit, F.M.; Zhang, M. Magnetite nanoparticles for medical MR imaging. Mater. Today 2011, 14, 330–338. [Google Scholar] [CrossRef]
- Hamzah, J.; Jugold, M.; Kiessling, F.; Rigby, P.; Manzur, M.; Marti, H.H.; Rabie, T.; Kaden, S.; Grone, H.J.; Hammerling, G.J.; et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 2008, 453, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Johansson-Percival, A.; He, B.; Li, Z.J.; Kjellen, A.; Russell, K.; Li, J.; Larma, I.; Ganss, R. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat. Immunol. 2017, 18, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Hellebust, A.; Richards-Kortum, R. Advances in molecular imaging: Targeted optical contrast agents for cancer diagnostics. Nanomedicine 2012, 7, 429–445. [Google Scholar] [CrossRef] [Green Version]
- Andreou, C.; Neuschmelting, V.; Tschaharganeh, D.F.; Huang, C.H.; Oseledchyk, A.; Iacono, P.; Karabeber, H.; Colen, R.R.; Mannelli, L.; Lowe, S.W.; et al. Imaging of liver tumors using surface-enhanced raman scattering nanoparticles. ACS Nano 2016, 10, 5015–5026. [Google Scholar] [CrossRef]
- Unterrainer, M.; Eze, C.; Ilhan, H.; Marschner, S.; Roengvoraphoj, O.; Schmidt-Hegemann, N.S.; Walter, F.; Kunz, W.G.; P. Rosenschold, M.A.; Jeraj, R.; et al. Recent advances of PET imaging in clinical radiation oncology. Radiat. Oncol. 2020, 15, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durymanov, M.O.; Rosenkranz, A.A.; Sobolev, A.S. Current approaches for improving intratumoral accumulation and distribution of nanomedicines. Theranostics 2015, 5, 1007–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, R.K.; Korn, R.L.; Raghunand, N.; Sachdev, J.C.; Newbold, R.G.; Jameson, G.; Fetterly, G.J.; Prey, J.; Klinz, S.G.; Kim, J.; et al. Correlation between ferumoxytol uptake in tumor lesions by MRI and response to nanoliposomal irinotecan in patients with advanced solid tumors: A pilot study. Clin. Cancer Res. 2017, 23, 3638–3648. [Google Scholar] [CrossRef] [Green Version]
- Borsi, L.; Balza, E.; Carnemolla, B.; Sassi, F.; Castellani, P.; Berndt, A.; Kosmehl, H.; Biro, A.; Siri, A.; Orecchia, P.; et al. Selective targeted delivery of TNFalpha to tumor blood vessels. Blood 2003, 102, 4384–4392. [Google Scholar] [CrossRef] [PubMed]
- Dakhel, S.; Ongaro, T.; Gouyou, B.; Matasci, M.; Villa, A.; Neri, D.; Cazzamalli, S. Targeted enhancement of the therapeutic window of L19-TNF by transient and selective inhibition of RIPK1-signaling cascade. Oncotarget 2019, 10, 6678–6690. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ring | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| d (Å) | 4.88 | 2.96 | 2.52 | 2.08 | 1.68 | 1.62 | 1.47 | 1.26 |
| hkl | 111 | 220 | 311 | 400 | 422 | 511 | 440 | 533 |
| NPs | Size (nm) | Zeta Potentials (mv) |
|---|---|---|
| Untargeted FAM-IO NP | 28.31 ± 4.77 | 32.20 ± 2.26 |
| FAM-CREKA-IO-NP | 35.40 ± 8.10 | 38.43 ± 7.10 |
| FAM-CSG-IO-NP | 33.83 ± 5.57 | 34.88 ± 4.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chopra, M.; Wu, J.; Yeow, Y.L.; Winteringham, L.; Clemons, T.D.; Saunders, M.; Kotamraju, V.R.; Ganss, R.; Feindel, K.W.; Hamzah, J. Enhanced Detection of Desmoplasia by Targeted Delivery of Iron Oxide Nanoparticles to the Tumour-Specific Extracellular Matrix. Pharmaceutics 2021, 13, 1663. https://doi.org/10.3390/pharmaceutics13101663
Chopra M, Wu J, Yeow YL, Winteringham L, Clemons TD, Saunders M, Kotamraju VR, Ganss R, Feindel KW, Hamzah J. Enhanced Detection of Desmoplasia by Targeted Delivery of Iron Oxide Nanoparticles to the Tumour-Specific Extracellular Matrix. Pharmaceutics. 2021; 13(10):1663. https://doi.org/10.3390/pharmaceutics13101663
Chicago/Turabian StyleChopra, Meenu, Jiansha Wu, Yen Ling Yeow, Louise Winteringham, Tristan D. Clemons, Martin Saunders, Venkata Ramana Kotamraju, Ruth Ganss, Kirk W. Feindel, and Juliana Hamzah. 2021. "Enhanced Detection of Desmoplasia by Targeted Delivery of Iron Oxide Nanoparticles to the Tumour-Specific Extracellular Matrix" Pharmaceutics 13, no. 10: 1663. https://doi.org/10.3390/pharmaceutics13101663
APA StyleChopra, M., Wu, J., Yeow, Y. L., Winteringham, L., Clemons, T. D., Saunders, M., Kotamraju, V. R., Ganss, R., Feindel, K. W., & Hamzah, J. (2021). Enhanced Detection of Desmoplasia by Targeted Delivery of Iron Oxide Nanoparticles to the Tumour-Specific Extracellular Matrix. Pharmaceutics, 13(10), 1663. https://doi.org/10.3390/pharmaceutics13101663