
Antibacterial Activity of T22, a Specific Peptidic Ligand of the Tumoral Marker CXCR4

 ,
,  , , , and
, , , and 
Abstract
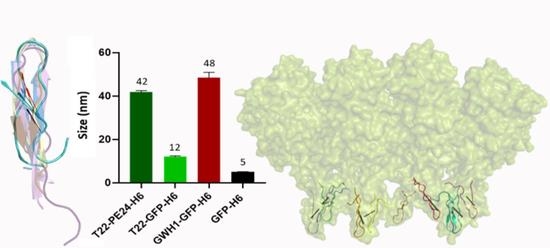
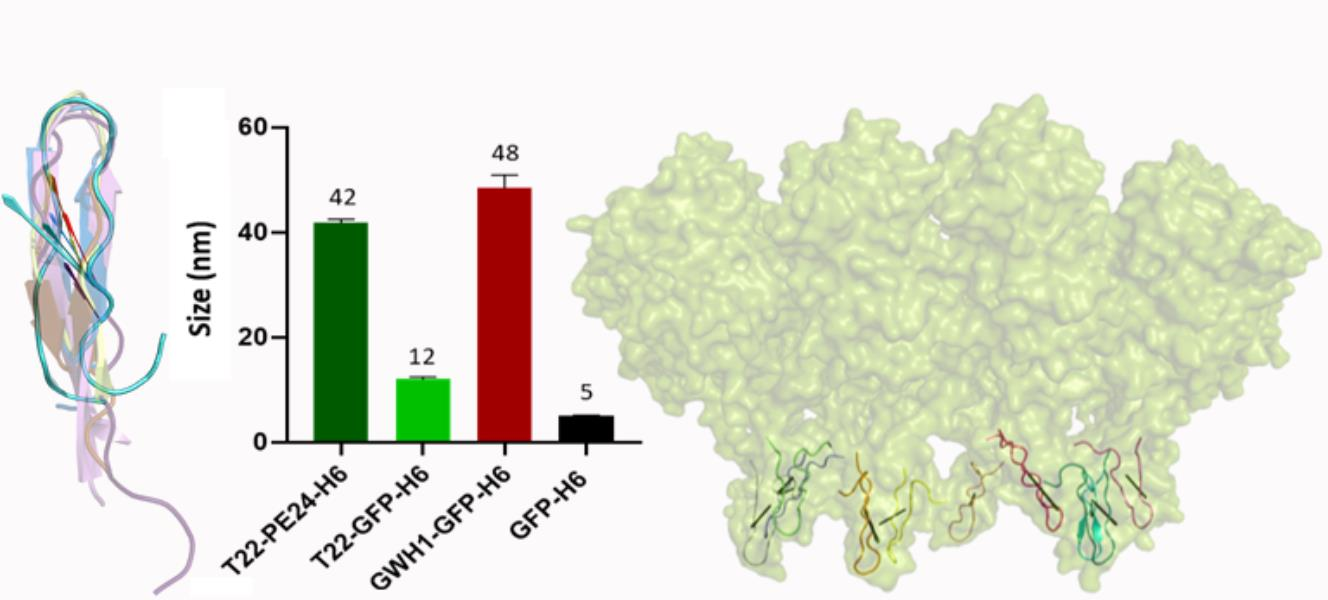
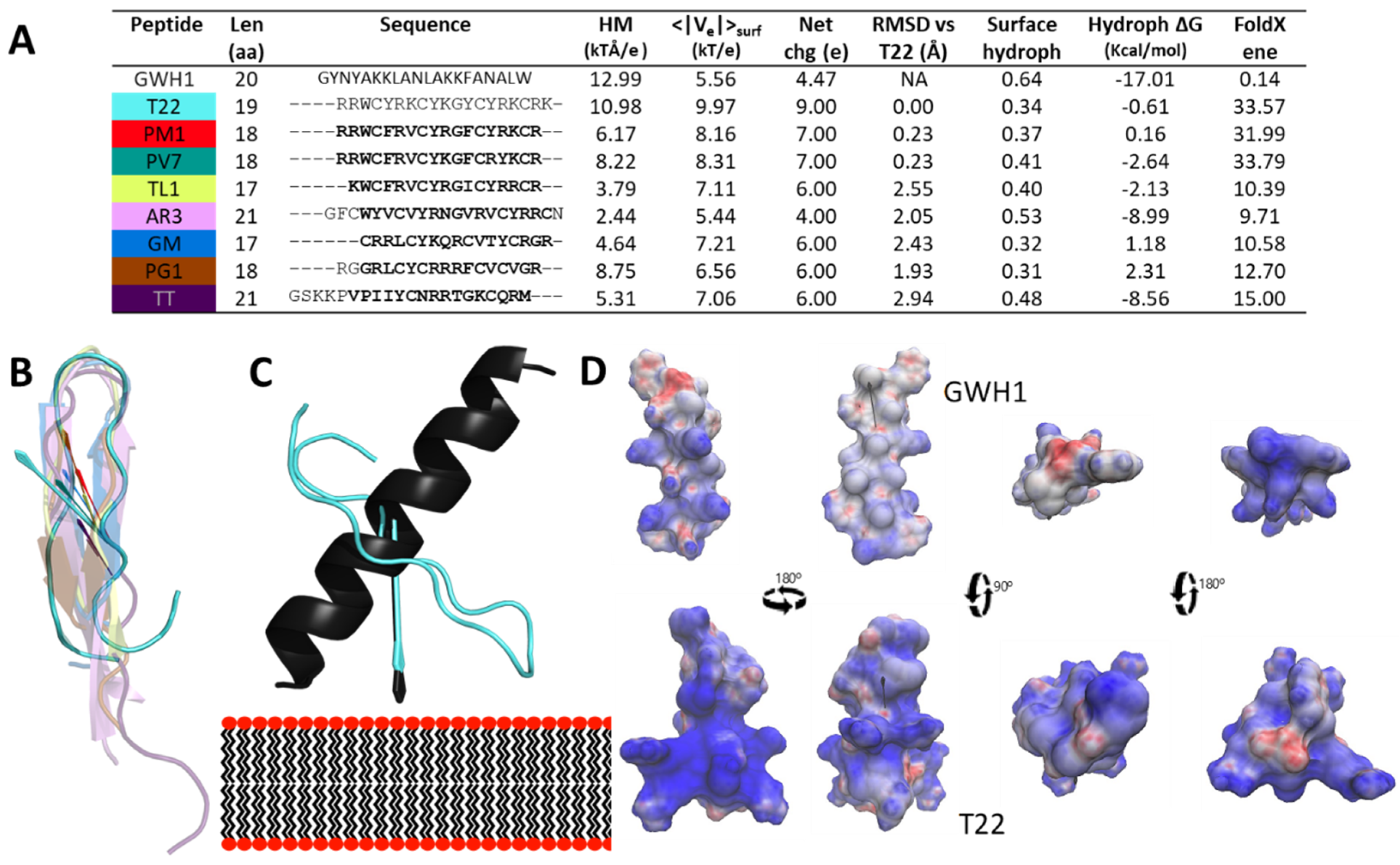
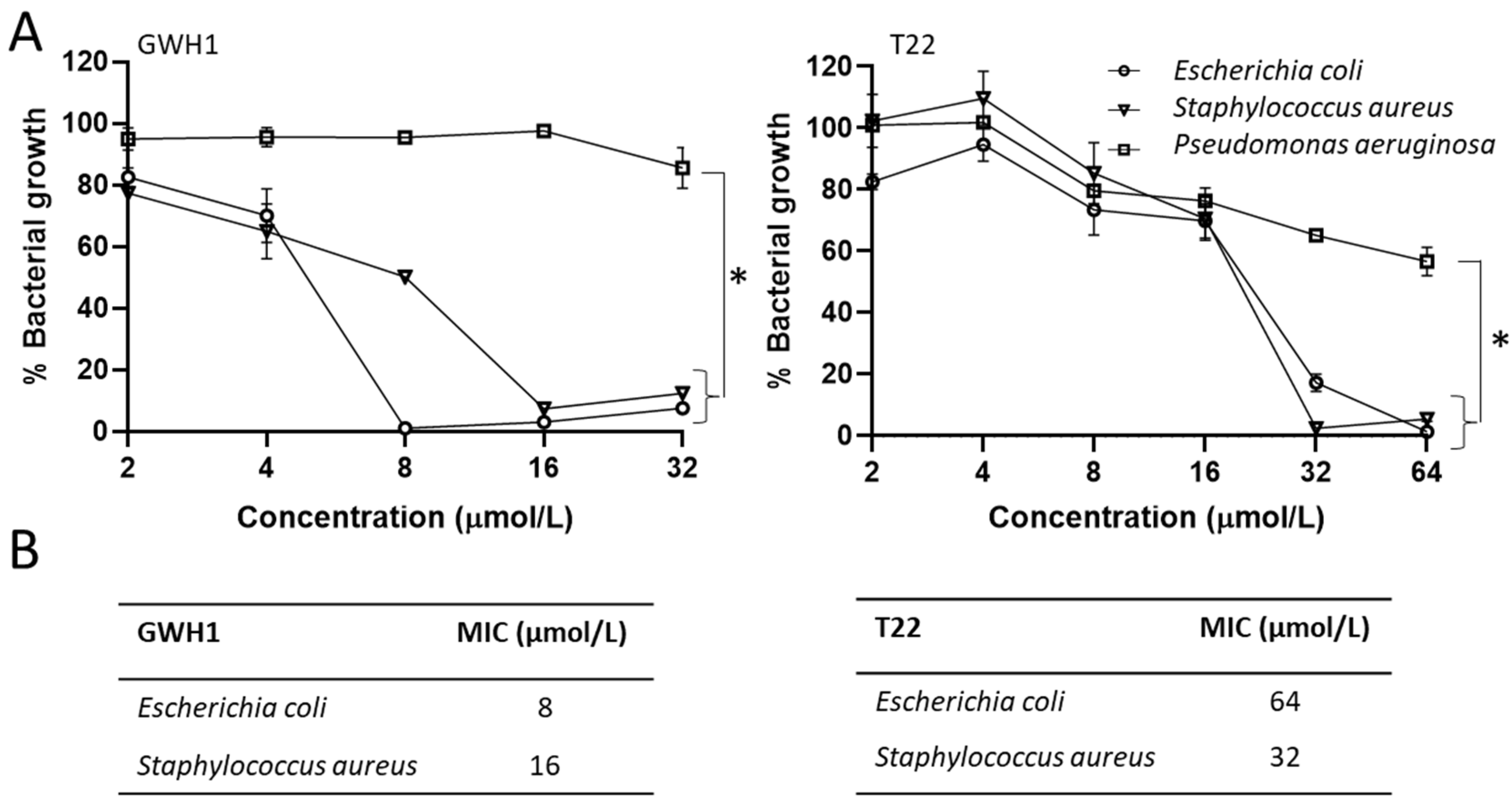
:
{kind=link}
{kind=link}
{kind=link}
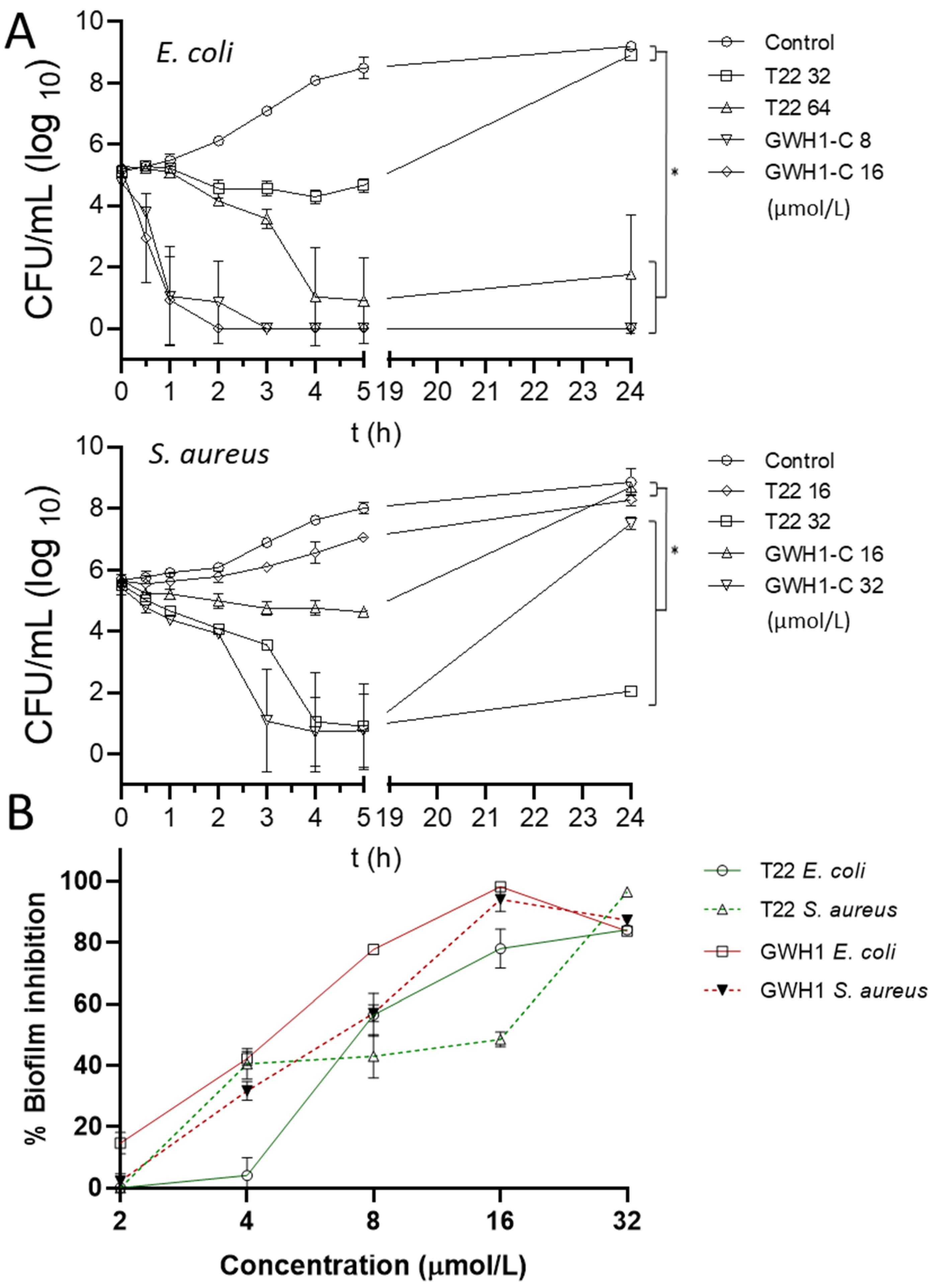{kind=link}
{kind=link}
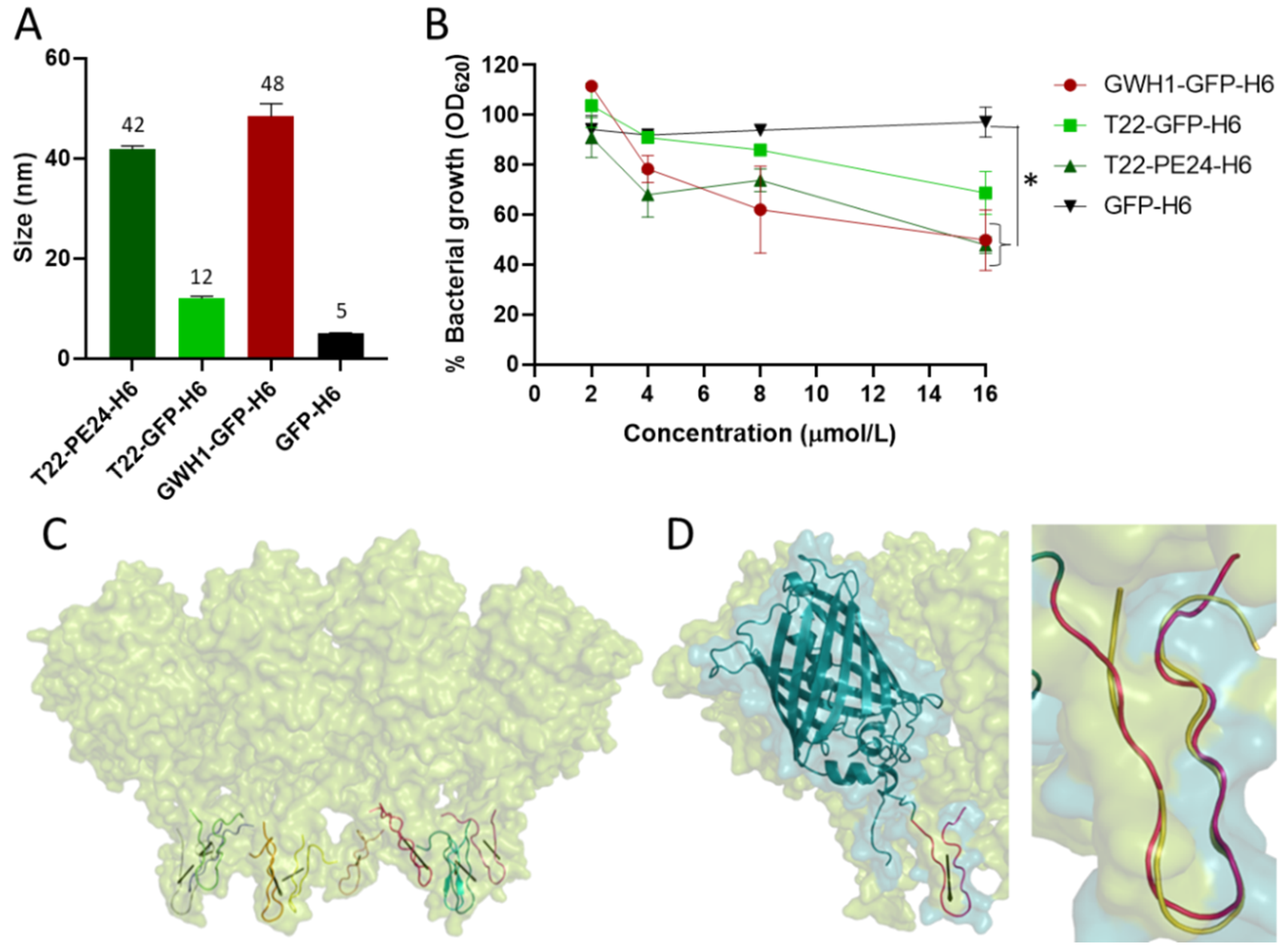{kind=link}
1. Introduction
2. Material and Methods
2.1. Peptides, Proteins and Protein Nanoparticles
2.2. Bacterial Growth and Determination of the Minimum Inhibitory Concentration
2.3. Time-Killing Kinetic Assay
2.4. Evaluation of Biofilm Formation
2.5. Mammalian Cell Viability Assay
2.6. Hemolysis Assay
2.7. Measurement of the Nanoparticle Size
2.8. Structure-Based Calculations and Molecular Modeling
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tamamura, H.; Arakaki, R.; Funakoshi, H.; Imai, M.; Otaka, A.; Ibuka, T.; Nakashima, H.; Murakami, T.; Waki, M.; Matsumoto, A.; et al. Effective lowly cytotoxic analogs of an HIV-cell fusion inhibitor, T22 ([Tyr5,12, Lys7]-polyphemusin II). Bioorg. Med. Chem. 1998, 6, 231–238. [Google Scholar] [CrossRef]
- Tamamura, H.; Imai, M.; Ishihara, T.; Masuda, M.; Funakoshi, H.; Oyake, H.; Murakami, T.; Arakaki, R.; Nakashima, H.; Otaka, A.; et al. Pharmacophore identification of a chemokine receptor (CXCR4) antagonist, T22 ([Tyr(5,12),Lys7]-polyphemusin II), which specifically blocks T cell-line-tropic HIV-1 infection. Bioorg. Med. Chem. 1998, 6, 1033–1041. [Google Scholar] [CrossRef]
- Zhou, N.; Luo, Z.; Luo, J.; Liu, D.; Hall, J.W.; Pomerantz, R.J.; Huang, Z. Structural and Functional Characterization of Human CXCR4 as a Chemokine Receptor and HIV-1 Co-receptor by Mutagenesis and Molecular Modeling Studies. J. Biol. Chem. 2001, 276, 42826–42833. [Google Scholar] [CrossRef] [Green Version]
- Tamamura, H.; Kuroda, M.; Masuda, M.; Otaka, A.; Funakoshi, S.; Nakashima, H.; Yamamoto, N.; Waki, M.; Matsumoto, A.; Lancelin, J.M.; et al. A comparative study of the solution structures of tachyplesin I and a novel anti-HIV synthetic peptide, T22 ([Tyr5,12, Lys7]-polyphemusin II), determined by nuclear magnetic resonance. Biochim. Biophys. Acta 1993, 1163, 209–216. [Google Scholar] [CrossRef]
- Casanova, I.; Parreno, M.; Farre, L.; Guerrero, S.; Cespedes, M.V.; Pavon, M.A.; Sancho, F.J.; Marcuello, E.; Trias, M.; Mangues, R. Celecoxib induces anoikis in human colon carcinoma cells associated with the deregulation of focal adhesions and nuclear translocation of p130Cas. Int. J. Cancer 2006, 118, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Cespedes, M.V.; Cano-Garrido, O.; Alamo, P.; Sala, R.; Gallardo, A.; Serna, N.; Falgas, A.; Volta-Duran, E.; Casanova, I.; Sanchez-Chardi, A.; et al. Engineering Secretory Amyloids for Remote and Highly Selective Destruction of Metastatic Foci. Adv. Mater. 2020, 32, 1907348. [Google Scholar] [CrossRef]
- Sanchez, J.M.; Lopez-Laguna, H.; Alamo, P.; Serna, N.; Sanchez-Chardi, A.; Nolan, V.; Cano-Garrido, O.; Casanova, I.; Unzueta, U.; Vazquez, E.; et al. Artificial Inclusion Bodies for Clinical Development. Adv. Sci. 2020, 7, 1902420. [Google Scholar] [CrossRef]
- Serna, N.; Alamo, P.; Ramesh, P.; Vinokurova, D.; Sanchez-Garcia, L.; Unzueta, U.; Gallardo, A.; Cespedes, M.V.; Vazquez, E.; Villaverde, A.; et al. Nanostructured toxins for the selective destruction of drug-resistant human CXCR4(+) colorectal cancer stem cells. J. Control. Release 2020, 320, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Falgas, A.; Pallares, V.; Unzueta, U.; Cespedes, M.V.; Arroyo-Solera, I.; Moreno, M.J.; Sierra, J.; Gallardo, A.; Mangues, M.A.; Vazquez, E.; et al. A CXCR4-targeted nanocarrier achieves highly selective tumor uptake in diffuse large B-cell lymphoma mouse models. Haematologica 2020, 105, 741–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falgas, A.; Pallares, V.; Serna, N.; Sanchez-Garcia, L.; Sierra, J.; Gallardo, A.; Alba-Castellon, L.; Alamo, P.; Unzueta, U.; Villaverde, A.; et al. Selective delivery of T22-PE24-H6 to CXCR4(+) diffuse large B-cell lymphoma cells leads to wide therapeutic index in a disseminated mouse model. Theranostics 2020, 10, 5169–5180. [Google Scholar] [CrossRef]
- Pallares, V.; Unzueta, U.; Falgas, A.; Sanchez-Garcia, L.; Serna, N.; Gallardo, A.; Morris, G.A.; Alba-Castellon, L.; Alamo, P.; Sierra, J.; et al. An Auristatin nanoconjugate targeting CXCR4+ leukemic cells blocks acute myeloid leukemia dissemination. J. Hematol. Oncol. 2020, 13, 36. [Google Scholar] [CrossRef] [Green Version]
- Rioja-Blanco, E.; Arroyo-Solera, I.; Álamo, P.; Casanova, I.; Gallardo, G.; Unzueta, U.; Serna, N.; Sánchez-García, L.; Quer, M.; Villaverde, A.; et al. Self-assembling protein nanocarrier for selective delivery of cytotoxic polypeptides to CXCR4+ head and neck squamous cell carcinoma tumors. Acta Pharm. Sinica B 2021, in press. [Google Scholar] [CrossRef]
- Sánchez-García, L.; Sala, R.; Serna, N.; Álamo, P.; Parladé, E.; Alba-Castellón, L.; Voltà-Durán, E.; Sánchez-Chardi, A.; Unzueta, U.; Vázquez, E.; et al. A refined cocktailing of pro-apoptotic nanoparticles boosts anti-tumor activity. Acta Biomater. 2020, 113, 584–596. [Google Scholar] [CrossRef]
- Serna, N.; Cano-Garrido, O.; Sánchez-García, L.; Pesarrodona, M.; Unzueta, U.; Sánchez-Chardi, A.; Mangues, R.; Vázquez, E.; Villaverde, A. Engineering Protein Venoms as Self-Assembling CXCR4-Targeted Cytotoxic Nanoparticles. Part. Part. Syst. Charact. 2020, 37, 2000040. [Google Scholar] [CrossRef]
- Unzueta, U.; Ferrer-Miralles, N.; Cedano, J.; Zikung, X.; Pesarrodona, M.; Saccardo, P.; Garcia-Fruitos, E.; Domingo-Espin, J.; Kumar, P.; Gupta, K.C.; et al. Non-amyloidogenic peptide tags for the regulatable self-assembling of protein-only nanoparticles. Biomaterials 2012, 33, 8714–8722. [Google Scholar] [CrossRef]
- Lopez-Laguna, H.; Unzueta, U.; Conchillo-Sole, O.; Sanchez-Chardi, A.; Pesarrodona, M.; Cano-Garrido, O.; Volta, E.; Sanchez-Garcia, L.; Serna, N.; Saccardo, P.; et al. Assembly of histidine-rich protein materials controlled through divalent cations. Acta Biomater. 2019, 83, 257–264. [Google Scholar] [CrossRef]
- Rueda, F.; Cespedes, M.V.; Conchillo-Sole, O.; Sanchez-Chardi, A.; Seras-Franzoso, J.; Cubarsi, R.; Gallardo, A.; Pesarrodona, M.; Ferrer-Miralles, N.; Daura, X.; et al. Bottom-Up Instructive Quality Control in the Biofabrication of Smart Protein Materials. Adv. Mater. 2015, 27, 7816–7822. [Google Scholar] [CrossRef] [Green Version]
- Cespedes, M.V.; Unzueta, U.; Avino, A.; Gallardo, A.; Alamo, P.; Sala, R.; Sanchez-Chardi, A.; Casanova, I.; Mangues, M.A.; Lopez-Pousa, A.; et al. Selective depletion of metastatic stem cells as therapy for human colorectal cancer. EMBO Mol. Med. 2018, 10, e8772. [Google Scholar] [CrossRef]
- Unzueta, U.; Roldan, M.; Pesarrodona, M.; Benitez, R.; Sanchez-Chardi, A.; Conchillo-Sole, O.; Mangues, R.; Villaverde, A.; Vazquez, E. Self-assembling as regular nanoparticles dramatically minimizes photobleaching of tumour-targeted GFP. Acta Biomater. 2020, 103, 272–280. [Google Scholar] [CrossRef]
- Powers, J.-P.S.; Tan, A.; Ramamoorthy, A.; Hancock, R.E.W. Solution Structure and Interaction of the Antimicrobial Polyphemusins with Lipid Membranes. Biochemistry 2005, 44, 15504–15513. [Google Scholar] [CrossRef] [Green Version]
- Ohta, M.; Ito, H.; Masuda, K.; Tanaka, S.; Arakawa, Y.; Wacharotayankun, R.; Kato, N. Mechanisms of antibacterial action of tachyplesins and polyphemusins, a group of antimicrobial peptides isolated from horseshoe crab hemocytes. Antimicrob. Agents Chemother. 1992, 36, 1460. [Google Scholar] [CrossRef] [Green Version]
- Marggraf, M.B.; Panteleev, P.V.; Emelianova, A.A.; Sorokin, M.I.; Bolosov, I.A.; Buzdin, A.A.; Kuzmin, D.V.; Ovchinnikova, T.V. Cytotoxic Potential of the Novel Horseshoe Crab Peptide Polyphemusin III. Mar. Drugs 2018, 16, 466. [Google Scholar] [CrossRef] [Green Version]
- Miyata, T.; Tokunaga, F.; Yoneya, T.; Yoshikawa, K.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Antimicrobial Peptides, Isolated from Horseshoe Crab Hemocytes, Tachyplesin II, and Polyphemusins I and II: Chemical Structures and Biological Activity1. J. Biochem. 1989, 106, 663–668. [Google Scholar] [CrossRef]
- Powers, J.-P.S.; Rozek, A.; Hancock, R.E.W. Structure–activity relationships for the β-hairpin cationic antimicrobial peptide polyphemusin I. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2004, 1698, 239–250. [Google Scholar] [CrossRef]
- Zhang, L.; Scott, M.G.; Yan, H.; Mayer, L.D.; Hancock, R.E.W. Interaction of Polyphemusin I and Structural Analogs with Bacterial Membranes, Lipopolysaccharide, and Lipid Monolayers. Biochemistry 2000, 39, 14504–14514. [Google Scholar] [CrossRef]
- Hollmann, A.; Martínez, M.; Noguera, M.E.; Augusto, M.T.; Disalvo, A.; Santos, N.C.; Semorile, L.; Maffía, P.C. Role of amphipathicity and hydrophobicity in the balance between hemolysis and peptide–membrane interactions of three related antimicrobial peptides. Colloids Surf. B Biointerfaces 2016, 141, 528–536. [Google Scholar] [CrossRef]
- Chen, C.H.; Starr, C.G.; Guha, S.; Wimley, W.C.; Ulmschneider, M.B.; Ulmschneider, J.P. Tuning of a Membrane-Perforating Antimicrobial Peptide to Selectively Target Membranes of Different Lipid Composition. J. Membr. Biol. 2021, 254, 75–96. [Google Scholar] [CrossRef]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.T.; Cooper, M.A. Contribution of Amphipathicity and Hydrophobicity to the Antimicrobial Activity and Cytotoxicity of β-Hairpin Peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef]
- Volta-Duran, E.; Serna, N.; Sanchez-Garcia, L.; Avino, A.; Sanchez, J.M.; Lopez-Laguna, H.; Cano-Garrido, O.; Casanova, I.; Mangues, R.; Eritja, R.; et al. Design and engineering of tumor-targeted, dual-acting cytotoxic nanoparticles. Acta Biomater. 2021, 119, 312–322. [Google Scholar] [CrossRef]
- Carratalá, J.V.; Serna, N.; Villaverde, A.; Vázquez, E.; Ferrer-Miralles, N. Nanostructured antimicrobial peptides: The last push towards clinics. Biotechnol. Adv. 2020, 44, 107603. [Google Scholar] [CrossRef]
- Carratala, J.V.; Brouillette, E.; Serna, N.; Sanchez-Chardi, A.; Sanchez, J.M.; Villaverde, A.; Aris, A.; Garcia-Fruitos, E.; Ferrer-Miralles, N.; Malouin, F. In Vivo Bactericidal Efficacy of GWH1 Antimicrobial Peptide Displayed on Protein Nanoparticles, a Potential Alternative to Antibiotics. Pharmaceutics 2020, 12, 1217. [Google Scholar] [CrossRef]
- van Elsland, D.; Neefjes, J. Bacterial infections and cancer. EMBO Rep. 2018, 19, e46632. [Google Scholar] [CrossRef]
- Eyvazi, S.; Vostakolaei, M.A.; Dilmaghani, A.; Borumandi, O.; Hejazi, M.S.; Kahroba, H.; Tarhriz, V. The oncogenic roles of bacterial infections in development of cancer. Microb. Pathog. 2020, 141, 104019. [Google Scholar] [CrossRef]
- Vogelmann, R.; Amieva, M.R. The role of bacterial pathogens in cancer. Curr. Opin. Microbiol. 2007, 10, 76–81. [Google Scholar] [CrossRef]
- Yadegarynia, D.; Tarrand, J.; Raad, I.; Rolston, K. Current Spectrum of Bacterial Infections in Patients with Cancer. Clin. Infect. Dis. 2003, 37, 1144–1145. [Google Scholar] [CrossRef]
- Michaud, D.S. Role of bacterial infections in pancreatic cancer. Carcinogenesis 2013, 34, 2193–2197. [Google Scholar] [CrossRef]
- Unzueta, U.; Cespedes, M.V.; Ferrer-Miralles, N.; Casanova, I.; Cedano, J.; Corchero, J.L.; Domingo-Espin, J.; Villaverde, A.; Mangues, R.; Vazquez, E. Intracellular CXCR4(+) cell targeting with T22-empowered protein-only nanoparticles. Int. J. Nanomed. 2012, 7, 4533–4544. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Garcia, L.; Serna, N.; Alamo, P.; Sala, R.; Cespedes, M.V.; Roldan, M.; Sanchez-Chardi, A.; Unzueta, U.; Casanova, I.; Mangues, R.; et al. Self-assembling toxin-based nanoparticles as self-delivered antitumoral drugs. J. Control. Release 2018, 274, 81–92. [Google Scholar] [CrossRef]
- Serna, N.; Sanchez-Garcia, L.; Sanchez-Chardi, A.; Unzueta, U.; Roldan, M.; Mangues, R.; Vazquez, E.; Villaverde, A. Protein-only, antimicrobial peptide-containing recombinant nanoparticles with inherent built-in antibacterial activity. Acta Biomater. 2017, 60, 256–263. [Google Scholar] [CrossRef]
- Vazquez, E.; Roldan, M.; Diez-Gil, C.; Unzueta, U.; Domingo-Espin, J.; Cedano, J.; Conchillo, O.; Ratera, I.; Veciana, J.; Daura, X.; et al. Protein nanodisk assembling and intracellular trafficking powered by an arginine-rich (R9) peptide. Nanomedicine 2010, 5, 259–268. [Google Scholar] [CrossRef]
- Haney, E.F.; Trimble, M.J.; Cheng, J.T.; Vallé, Q.; Hancock, R.E.W. Critical Assessment of Methods to Quantify Biofilm Growth and Evaluate Antibiofilm Activity of Host Defence Peptides. Biomolecules 2018, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Elliott, A.G.; Huang, J.X.; Neve, S.; Zuegg, J.; Edwards, I.A.; Cain, A.K.; Boinett, C.J.; Barquist, L.; Lundberg, C.V.; Steen, J.; et al. An amphipathic peptide with antibiotic activity against multidrug-resistant Gram-negative bacteria. Nat. Commun. 2020, 11, 3184. [Google Scholar] [CrossRef]
- Fahrner, R.L.; Dieckmann, T.; Harwig, S.S.L.; Lehrer, R.I.; Eisenberg, D.; Feigon, J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996, 3, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Mandard, N.; Bulet, P.; Caille, A.; Daffre, S.; Vovelle, F. The solution structure of gomesin, an antimicrobial cysteine-rich peptide from the spider. Eur. J. Biochem. 2002, 269, 1190–1198. [Google Scholar] [CrossRef]
- Mandard, N.; Sodano, P.; Labbe, H.; Bonmatin, J.-M.; Bulet, P.; Hetru, C.; Ptak, M.; Vovelle, F. Solution structure of thanatin, a potent bactericidal and fungicidal insect peptide, determined from proton two-dimensional nuclear magnetic resonance data. Eur. J. Biochem. 1998, 256, 404–410. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- Reißer, S.; Strandberg, E.; Steinbrecher, T.; Ulrich, A.S. 3D Hydrophobic Moment Vectors as a Tool to Characterize the Surface Polarity of Amphiphilic Peptides. Biophys. J. 2014, 106, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Nagarajan, R.; Archana, A.; Thangakani, A.M.; Jemimah, S.; Velmurugan, D.; Gromiha, M.M. PDBparam: Online Resource for Computing Structural Parameters of Proteins. Bioinform. Biol. Insights 2016, 10, 73–80. [Google Scholar] [CrossRef] [Green Version]
- McLachlan, A.D. Rapid comparison of protein structures. Acta Crystallogr. Sect. A 1982, 38, 871–873. [Google Scholar] [CrossRef]
- Shindyalov, I.N.; Bourne, P.E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. Des. Sel. 1998, 11, 739–747. [Google Scholar] [CrossRef]
- Chen, Y.L.; Li, J.H.; Yu, C.Y.; Lin, C.J.; Chiu, P.H.; Chen, P.W.; Lin, C.C.; Chen, W.J. Novel cationic antimicrobial peptide GW-H1 induced caspase-dependent apoptosis of hepatocellular carcinoma cell lines. Peptides 2012, 36, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.T.; Kuo, T.Y.; Chiang, J.C.; Pei, M.J.; Yang, W.T.; Yu, H.C.; Lin, S.B.; Chen, W.J. Design and synthesis of cationic antimicrobial peptides with improved activity and selectivity against Vibrio spp. Int. J. Antimicrob. Agents 2008, 32, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Serna, N.; Carratalá, J.V.; Parladé, E.; Sánchez-Chardi, A.; Aviñó, A.; Unzueta, U.; Mangues, R.; Eritja, R.; Ferrer-Miralles, N.; Vazquez, E.; et al. Developing Protein–Antitumoral Drug Nanoconjugates as Bifunctional Antimicrobial Agents. ACS Appl. Mater. Interfaces 2020, 12, 57746–57756. [Google Scholar] [CrossRef]
- Greco, I.; Molchanova, N.; Holmedal, E.; Jenssen, H.; Hummel, B.D.; Watts, J.L.; Håkansson, J.; Hansen, P.R.; Svenson, J. Correlation between hemolytic activity, cytotoxicity and systemic in vivo toxicity of synthetic antimicrobial peptides. Sci. Rep. 2020, 10, 13206. [Google Scholar] [CrossRef]
- Maher, S.; McClean, S. Investigation of the cytotoxicity of eukaryotic and prokaryotic antimicrobial peptides in intestinal epithelial cells in vitro. Biochem. Pharmacol. 2006, 71, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.; Gudiol, C.; Garcia-Vidal, C.; Ardanuy, C.; Carratala, J. Bloodstream infections in patients with solid tumors: Epidemiology, antibiotic therapy, and outcomes in 528 episodes in a single cancer center. Medicine 2014, 93, 143–149. [Google Scholar] [CrossRef]
- Rolston, K.V. Infections in Cancer Patients with Solid Tumors: A Review. Infect. Dis. Ther. 2017, 6, 69–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolston, K.V. Polymicrobial pulmonary infections in cancer patients with underlying solid tumors. Infection 2017, 45, 245–246. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver–passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Gagniere, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Antonic, V.; Stojadinovic, A.; Kester, K.E.; Weina, P.J.; Brucher, B.L.; Protic, M.; Avital, I.; Izadjoo, M. Significance of infectious agents in colorectal cancer development. J. Cancer 2013, 4, 227–240. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the human colorectal cancer microbiome. PLoS ONE 2011, 6, e20447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaei, R.; Mirzaei, H.; Alikhani, M.Y.; Sholeh, M.; Arabestani, M.R.; Saidijam, M.; Karampoor, S.; Ahmadyousefi, Y.; Moghadam, M.S.; Irajian, G.R.; et al. Bacterial biofilm in colorectal cancer: What is the real mechanism of action? Microb. Pathog. 2020, 142, 104052. [Google Scholar] [CrossRef] [PubMed]
- Serna, R.; Sánchez-García, L.; Unzueta, U.; Díaz, R.; Vázquez, E.; Mangues, R.; Villaverde, A. Protein-based therapeutic killing for cancer therapies. Trends Biotechnol. 2018, 36, 318–335. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, C. Side effects related to systemic cancer treatment: Are we changing the Promethean experience with molecularly targeted therapies? Curr. Oncol. 2008, 15, 198–199. [Google Scholar] [CrossRef] [PubMed]
- Senkus, E.; Jassem, J. Cardiovascular effects of systemic cancer treatment. Cancer Treat. Rev. 2011, 37, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Pearce, A.; Haas, M.; Viney, R.; Pearson, S.A.; Haywood, P.; Brown, C.; Ward, R. Incidence and severity of self-reported chemotherapy side effects in routine care: A prospective cohort study. PLoS ONE 2017, 12, e0184360. [Google Scholar] [CrossRef]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Casanova, I.; Unzueta, U.; Arroyo-Solera, I.; Cespedes, M.V.; Villaverde, A.; Mangues, R.; Vazquez, E. Protein-driven nanomedicines in oncotherapy. Curr. Opin. Pharmacol. 2019, 47, 1–7. [Google Scholar] [CrossRef]
- Tran, S.; DeGiovanni, P.J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Yao, V.J.; D’Angelo, S.; Butler, K.S.; Theron, C.; Smith, T.L.; Marchio, S.; Gelovani, J.G.; Sidman, R.L.; Dobroff, A.S.; Brinker, C.J.; et al. Ligand-targeted theranostic nanomedicines against cancer. J. Control. Release 2016, 240, 267–286. [Google Scholar] [CrossRef] [Green Version]
- Gener, P.; Rafael, D.F.; Fernandez, Y.; Ortega, J.S.; Arango, D.; Abasolo, I.; Videira, M.; Schwartz, S., Jr. Cancer stem cells and personalized cancer nanomedicine. Nanomedicine 2016, 11, 307–320. [Google Scholar] [CrossRef]
- Kulbe, H.; Levinson, N.R.; Balkwill, F.; Wilson, J.L. The chemokine network in cancer--much more than directing cell movement. Int. J. Dev. Biol. 2004, 48, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Cardones, A.R.; Hwang, S.T. Chemokine receptors and melanoma metastasis. J. Dermatol. Sci. 2004, 36, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Takeuchi, H.; Lam, S.T.; Turner, R.R.; Wang, H.J.; Kuo, C.; Foshag, L.; Bilchik, A.J.; Hoon, D.S. Chemokine receptor CXCR4 expression in colorectal cancer patients increases the risk for recurrence and for poor survival. J. Clin. Oncol. 2005, 23, 2744–2753. [Google Scholar] [CrossRef]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef]
- Kim, J.; Mori, T.; Chen, S.L.; Amersi, F.F.; Martinez, S.R.; Kuo, C.; Turner, R.R.; Ye, X.; Bilchik, A.J.; Morton, D.L.; et al. Chemokine receptor CXCR4 expression in patients with melanoma and colorectal cancer liver metastases and the association with disease outcome. Ann. Surg. 2006, 244, 113–120. [Google Scholar] [CrossRef]
- Croker, A.K.; Allan, A.L. Cancer stem cells: Implications for the progression and treatment of metastatic disease. J. Cell. Mol. Med. 2008, 12, 374–390. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, F.; Bajetto, A.; Florio, T. Role of chemokine network in the development and progression of ovarian cancer: A potential novel pharmacological target. J. Oncol. 2010, 2010, 426956. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kang, D.H.; Chung, D.Y.; Kwon, J.K.; Lee, H.; Cho, N.H.; Choi, Y.D.; Hong, S.J.; Cho, K.S. Meta-Analysis of the Relationship between CXCR4 Expression and Metastasis in Prostate Cancer. World J. Mens Health 2014, 32, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Ni, C.; Chen, W.; Wu, P.; Wang, Z.; Yin, J.; Huang, J.; Qiu, F. Expression of CXCR4 and breast cancer prognosis: A systematic review and meta-analysis. BMC Cancer 2014, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.F.; Liu, S.Y.; Min, X.Y.; Ji, Y.Y.; Wang, N.; Liu, D.; Ma, N.; Li, Z.F.; Li, K. The prognostic value of CXCR4 in ovarian cancer: A meta-analysis. PLoS ONE 2014, 9, e92629. [Google Scholar] [CrossRef] [Green Version]
- Moreno, M.J.; Bosch, R.; Dieguez-Gonzalez, R.; Novelli, S.; Mozos, A.; Gallardo, A.; Pavon, M.A.; Cespedes, M.V.; Granena, A.; Alcoceba, M.; et al. CXCR4 expression enhances diffuse large B cell lymphoma dissemination and decreases patient survival. J. Pathol. 2015, 235, 445–455. [Google Scholar] [CrossRef]
- Salan, A.I.; Marasescu, P.C.; Camen, A.; Ciuca, E.M.; Matei, M.; Florescu, A.M.; Padureanu, V.; Margaritescu, C. The prognostic value of CXCR4, alpha-SMA and WASL in upper lip basal cell carcinomas. Rom. J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2018, 59, 839–849. [Google Scholar]
- Diaz, R.; Pallares, V.; Cano-Garrido, O.; Serna, N.; Sanchez-Garcia, L.; Falgas, A.; Pesarrodona, M.; Unzueta, U.; Sanchez-Chardi, A.; Sanchez, J.M.; et al. Selective CXCR4(+) Cancer Cell Targeting and Potent Antineoplastic Effect by a Nanostructured Version of Recombinant Ricin. Small 2018, 14, e1800665. [Google Scholar] [CrossRef] [PubMed]
- Álamo, P.; Parladé, P.; López-Laguna, H.; Voltà-Durán, E.; Unzueta, U.; Vazquez, E.; Mangues, R.; Villaverde, A. Ion-dependent slow protein release from in vivo disintegrating micro-granules. Drug Deliv. 2021, 28, 2383–2391. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cao, F.; Zhang, G.; Shi, L.; Chen, S.; Zhang, Z.; Zhi, W.; Ma, T. Trends in and Predictions of Colorectal Cancer Incidence and Mortality in China From 1990 to 2025. Front. Oncol. 2019, 9, 98. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.S. Social determinants of colorectal cancer risk, stage, and survival: A systematic review. Int. J. Colorectal Dis. 2020, 35, 985–995. [Google Scholar] [CrossRef]
- Kudo, S.; Lambert, R.; Allen, J.I.; Fujii, H.; Fujii, T.; Kashida, H.; Matsuda, T.; Mori, M.; Saito, H.; Shimoda, T.; et al. Nonpolypoid neoplastic lesions of the colorectal mucosa. Gastrointest. Endosc. 2008, 68, S3–S47. [Google Scholar] [CrossRef]
- Kudo, S.; Kashida, H.; Tamura, T.; Kogure, E.; Imai, Y.; Yamano, H.; Hart, A.R. Colonoscopic diagnosis and management of nonpolypoid early colorectal cancer. World J. Surg. 2000, 24, 1081–1090. [Google Scholar] [CrossRef]
- Kudo, S.; Kashida, H.; Nakajima, T.; Tamura, S.; Nakajo, K. Endoscopic diagnosis and treatment of early colorectal cancer. World J. Surg. 1997, 21, 694–701. [Google Scholar] [CrossRef]
- Hurlstone, D.P.; Cross, S.S.; Drew, K.; Adam, I.; Shorthouse, A.J.; Brown, S.; Sanders, D.S.; Lobo, A.J. An evaluation of colorectal endoscopic mucosal resection using high-magnification chromoscopic colonoscopy: A prospective study of 1000 colonoscopies. Endoscopy 2004, 36, 491–498. [Google Scholar] [CrossRef]
- Kim, M.N.; Kang, J.M.; Yang, J.I.; Kim, B.K.; Im, J.P.; Kim, S.G.; Jung, H.C.; Song, I.S.; Kim, J.S. Clinical features and prognosis of early colorectal cancer treated by endoscopic mucosal resection. J. Gastroenterol. Hepatol. 2011, 26, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, T.; Zhang, L.; Liu, X.J.; Xu, J.M.; Bai, Y.X.; Wang, Y.; Han, Y.; Li, Y.H.; Ba, Y. Bevacizumab plus irinotecan, 5-fluorouracil, and leucovorin (FOLFIRI) as the second-line therapy for patients with metastatic colorectal cancer, a multicenter study. Med. Oncol. 2013, 30, 752. [Google Scholar] [CrossRef]
- Iwasa, S.; Nagashima, K.; Yamaguchi, T.; Matsumoto, H.; Ichikawa, Y.; Goto, A.; Yasui, H.; Kato, K.; Okita, N.T.; Shimada, Y.; et al. S-1 and irinotecan with or without bevacizumab versus 5-fluorouracil and leucovorin plus oxaliplatin with or without bevacizumab in metastatic colorectal cancer: A pooled analysis of four phase II studies. Cancer Chemother. Pharmacol. 2015, 76, 605–614. [Google Scholar] [CrossRef]
- Xu, R.H.; Muro, K.; Morita, S.; Iwasa, S.; Han, S.W.; Wang, W.; Kotaka, M.; Nakamura, M.; Ahn, J.B.; Deng, Y.H.; et al. Modified XELIRI (capecitabine plus irinotecan) versus FOLFIRI (leucovorin, fluorouracil, and irinotecan), both either with or without bevacizumab, as second-line therapy for metastatic colorectal cancer (AXEPT): A multicentre, open-label, randomised, non-inferiority, phase 3 trial. Lancet Oncol. 2018, 19, 660–671. [Google Scholar] [CrossRef]
- Hu, J.; Chen, C.; Zhang, S.; Zhao, X.; Xu, H.; Zhao, X.; Lu, J.R. Designed antimicrobial and antitumor peptides with high selectivity. Biomacromolecules 2011, 12, 3839–3843. [Google Scholar] [CrossRef]
- Yang, C.H.; Chen, Y.C.; Peng, S.Y.; Tsai, A.P.; Lee, T.J.; Yen, J.H.; Liou, J.W. An engineered arginine-rich alpha-helical antimicrobial peptide exhibits broad-spectrum bactericidal activity against pathogenic bacteria and reduces bacterial infections in mice. Sci. Rep. 2018, 8, 14602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedron, C.N.; de Oliveira, C.S.; da Silva, A.F.; Andrade, G.P.; da Silva Pinhal, M.A.; Cerchiaro, G.; da Silva Junior, P.I.; da Silva, F.D.; Torres, M.T.; Oliveira, V.X. The effect of lysine substitutions in the biological activities of the scorpion venom peptide VmCT1. Eur. J. Pharm. Sci. 2019, 136, 104952. [Google Scholar] [CrossRef]
- Mattei, B.; Miranda, A.; Perez, K.R.; Riske, K.A. Structure–Activity Relationship of the Antimicrobial Peptide Gomesin: The Role of Peptide Hydrophobicity in Its Interaction with Model Membranes. Langmuir 2014, 30, 3513–3521. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Bentz, D.; Wadhwani, P.; Bürck, J.; Ulrich, A.S. Terminal charges modulate the pore forming activity of cationic amphipathic helices. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183243. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.N.; Rajpoot, K.; K Jain, S.K.J. Using 5-fluorouracil-encored plga nanoparticles for the treatment of colorectal cancer: The in-vitro characterization and cytotoxicity studies. Nanomedicine J. 2020, 7, 211–224. [Google Scholar] [CrossRef]
- Feng, C.; Li, J.; Kong, M.; Liu, Y.; Cheng, X.J.; Li, Y.; Park, H.J.; Chen, X.G. Surface charge effect on mucoadhesion of chitosan based nanogels for local anti-colorectal cancer drug delivery. Colloids Surf. B Biointerfaces 2015, 128, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Pridgen, E.M.; Alexis, F.; Farokhzad, O.C. Polymeric nanoparticle drug delivery technologies for oral delivery applications. Expert Opin. Drug Deliv. 2015, 12, 1459–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wathoni, N.; Nguyen, A.N.; Rusdin, A.; Umar, A.K.; Mohammed, A.F.A.; Motoyama, K.; Joni, I.M.; Muchtaridi, M. Enteric-Coated Strategies in Colorectal Cancer Nanoparticle Drug Delivery System. Drug Des. Dev. Ther. 2020, 14, 4387–4405. [Google Scholar] [CrossRef] [PubMed]
- Coco, R.; Plapied, L.; Pourcelle, V.; Jérôme, C.; Brayden, D.J.; Schneider, Y.-J.; Préat, V. Drug delivery to inflamed colon by nanoparticles: Comparison of different strategies. Int. J. Pharm. 2013, 440, 3–12. [Google Scholar] [CrossRef]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Peppelenbosch, M.P.; Smits, R. Bacterial biofilms as a potential contributor to mucinous colorectal cancer formation. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2019, 1872, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Konstantinov, S.R.; Smits, R.; Peppelenbosch, M.P. Bacterial Biofilms in Colorectal Cancer Initiation and Progression. Trends Mol. Med. 2017, 23, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Drewes, J.L.; White, J.R.; Dejea, C.M.; Fathi, P.; Iyadorai, T.; Vadivelu, J.; Roslani, A.C.; Wick, E.C.; Mongodin, E.F.; Loke, M.F.; et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microbiomes 2017, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Zarei, O.; Arabestan, M.R.; Majlesi, A.; Mohammadi, Y.; Alikhani, M.Y. Determination of virulence determinants of Escherichia coli strains isolated from patients with colorectal cancer compared to the healthy subjects. Gastroenterol. Hepatol. Bed Bench 2019, 12, 52–59. [Google Scholar]
- Kosari, F.; Taheri, M.; Moradi, A.; Hakimi Alni, R.; Alikhani, M.Y. Evaluation of cinnamon extract effects on clbB gene expression and biofilm formation in Escherichia coli strains isolated from colon cancer patients. BMC Cancer 2020, 20, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Castro, E.J.T.; Shim, H.; Advincula, J.V.G.; Kim, Y.-W. Differences Regarding the Molecular Features and Gut Microbiota Between Right and Left Colon Cancer. Ann. Coloproctol. 2018, 34, 280–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.; Menter, D.G.; Kopetz, S. Right Versus Left Colon Cancer Biology: Integrating the Consensus Molecular Subtypes. J. Natl. Compr. Cancer Netw. 2017, 15, 411–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serna, N.; Carratalá, J.V.; Conchillo-Solé, O.; Martínez-Torró, C.; Unzueta, U.; Mangues, R.; Ferrer-Miralles, N.; Daura, X.; Vázquez, E.; Villaverde, A. Antibacterial Activity of T22, a Specific Peptidic Ligand of the Tumoral Marker CXCR4. Pharmaceutics 2021, 13, 1922. https://doi.org/10.3390/pharmaceutics13111922
Serna N, Carratalá JV, Conchillo-Solé O, Martínez-Torró C, Unzueta U, Mangues R, Ferrer-Miralles N, Daura X, Vázquez E, Villaverde A. Antibacterial Activity of T22, a Specific Peptidic Ligand of the Tumoral Marker CXCR4. Pharmaceutics. 2021; 13(11):1922. https://doi.org/10.3390/pharmaceutics13111922
Chicago/Turabian StyleSerna, Naroa, José Vicente Carratalá, Oscar Conchillo-Solé, Carlos Martínez-Torró, Ugutz Unzueta, Ramón Mangues, Neus Ferrer-Miralles, Xavier Daura, Esther Vázquez, and Antonio Villaverde. 2021. "Antibacterial Activity of T22, a Specific Peptidic Ligand of the Tumoral Marker CXCR4" Pharmaceutics 13, no. 11: 1922. https://doi.org/10.3390/pharmaceutics13111922
APA StyleSerna, N., Carratalá, J. V., Conchillo-Solé, O., Martínez-Torró, C., Unzueta, U., Mangues, R., Ferrer-Miralles, N., Daura, X., Vázquez, E., & Villaverde, A. (2021). Antibacterial Activity of T22, a Specific Peptidic Ligand of the Tumoral Marker CXCR4. Pharmaceutics, 13(11), 1922. https://doi.org/10.3390/pharmaceutics13111922