
Evaluation of a Keratin 1 Targeting Peptide-Doxorubicin Conjugate in a Mouse Model of Triple-Negative Breast Cancer



Abstract
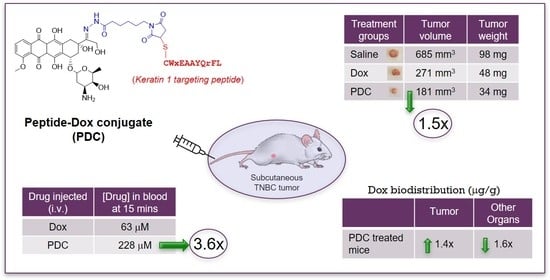
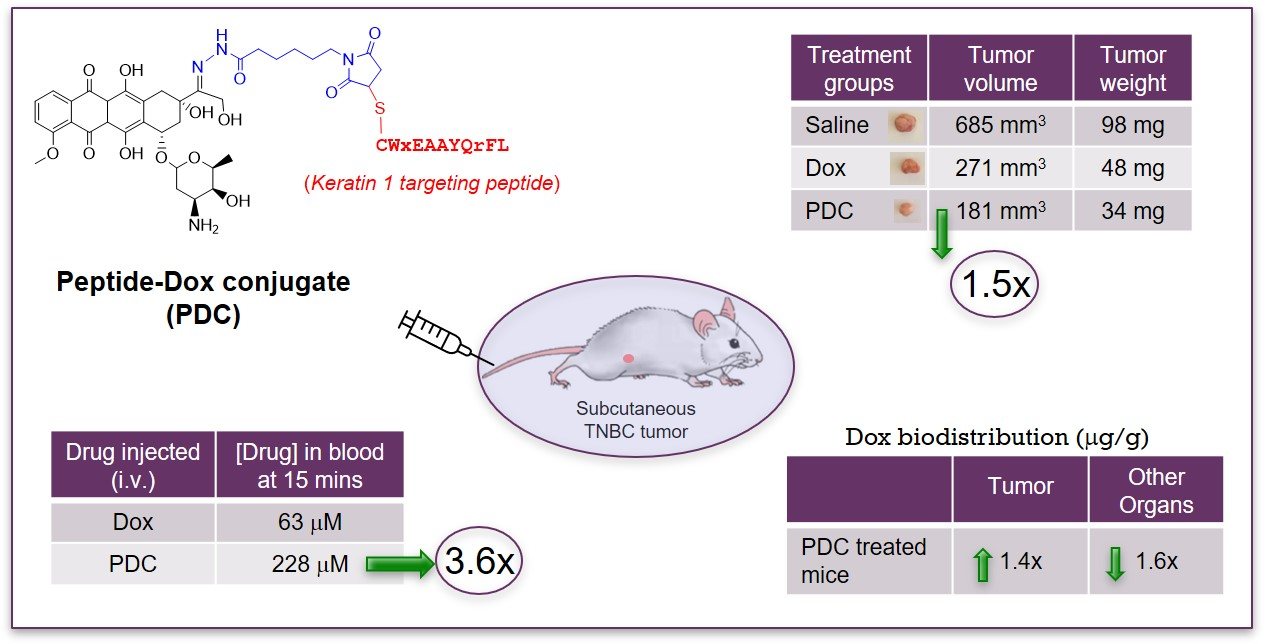
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Conjugate Solution for Intravenous Injections
2.2. Drug Circulation Time
2.3. Cell Culture and Tumor Inoculation
2.4. Drug Treatment
2.5. Biodistribution of Drug
2.6. Statistical Analysis
3. Results
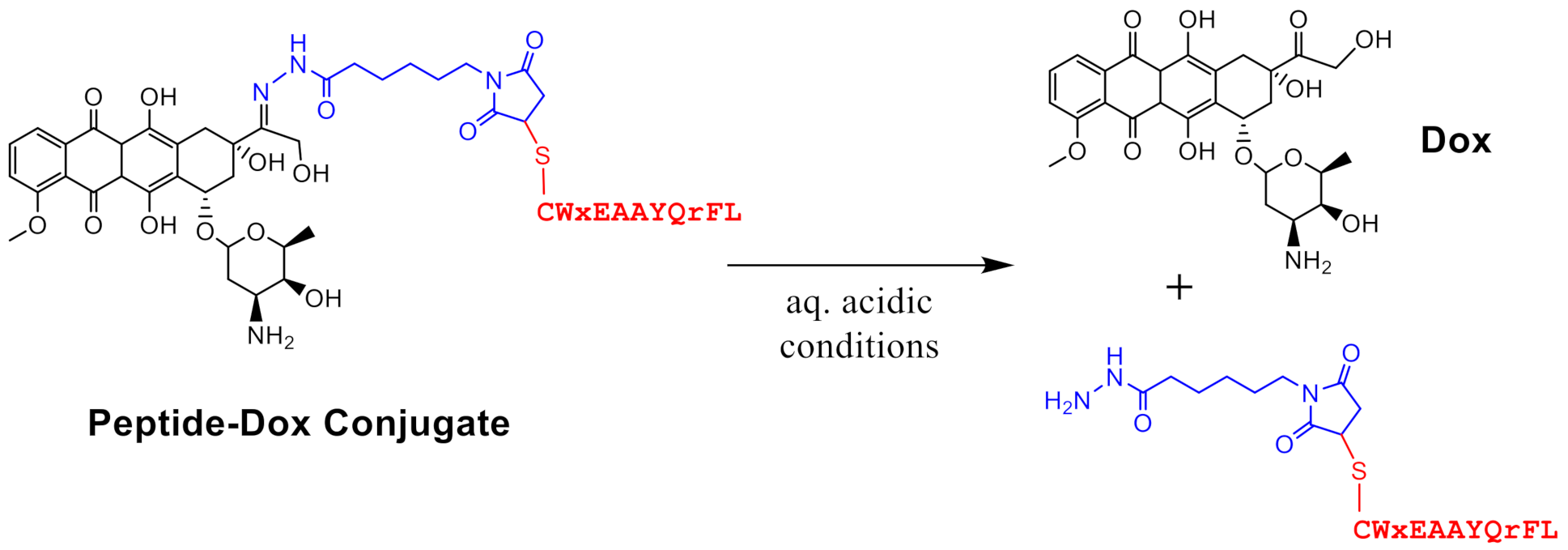
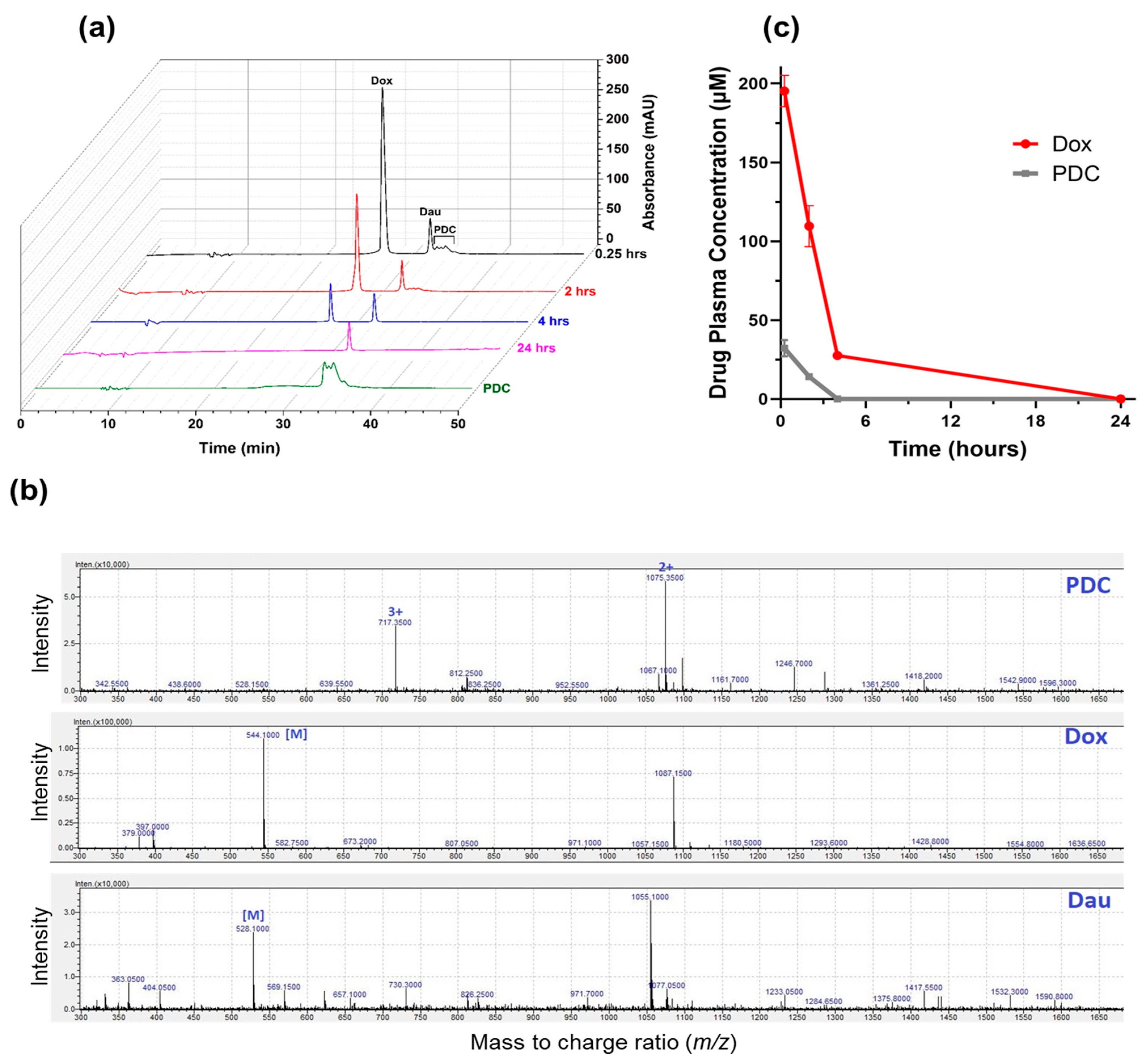
3.1. In Vivo Stability of Peptide-Dox Conjugate
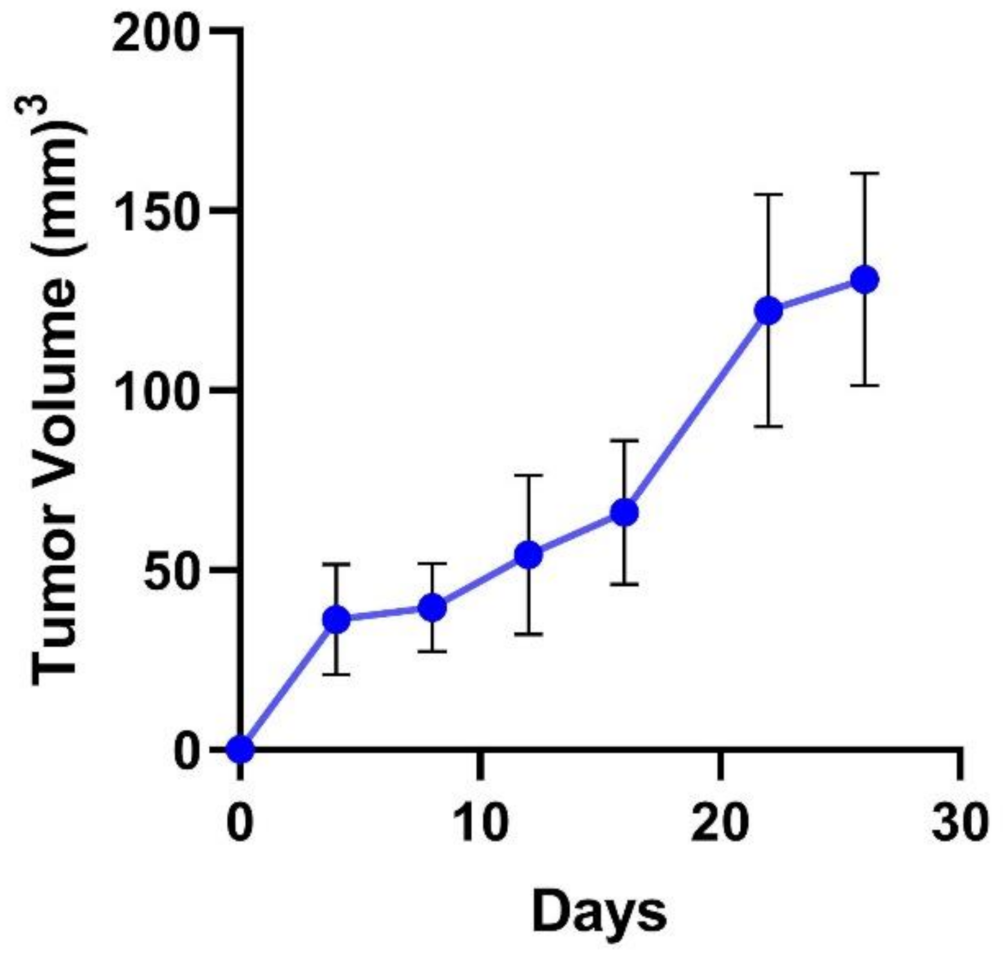
3.2. TNBC Tumor Growth Rate in a Subcutaneous Cell-Derived Xenograft Model
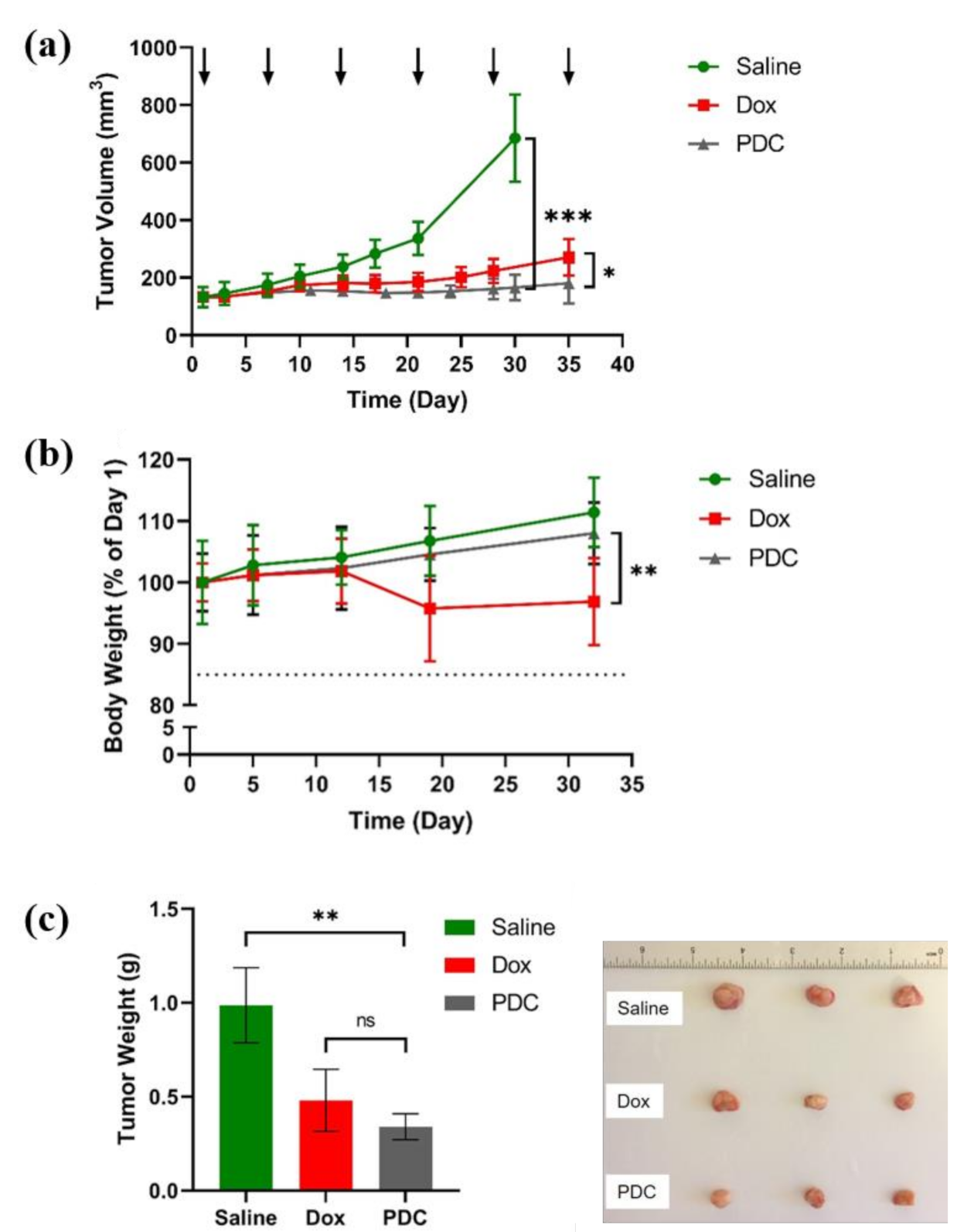
3.3. In Vivo Antitumor Effect of Peptide-Dox Conjugate
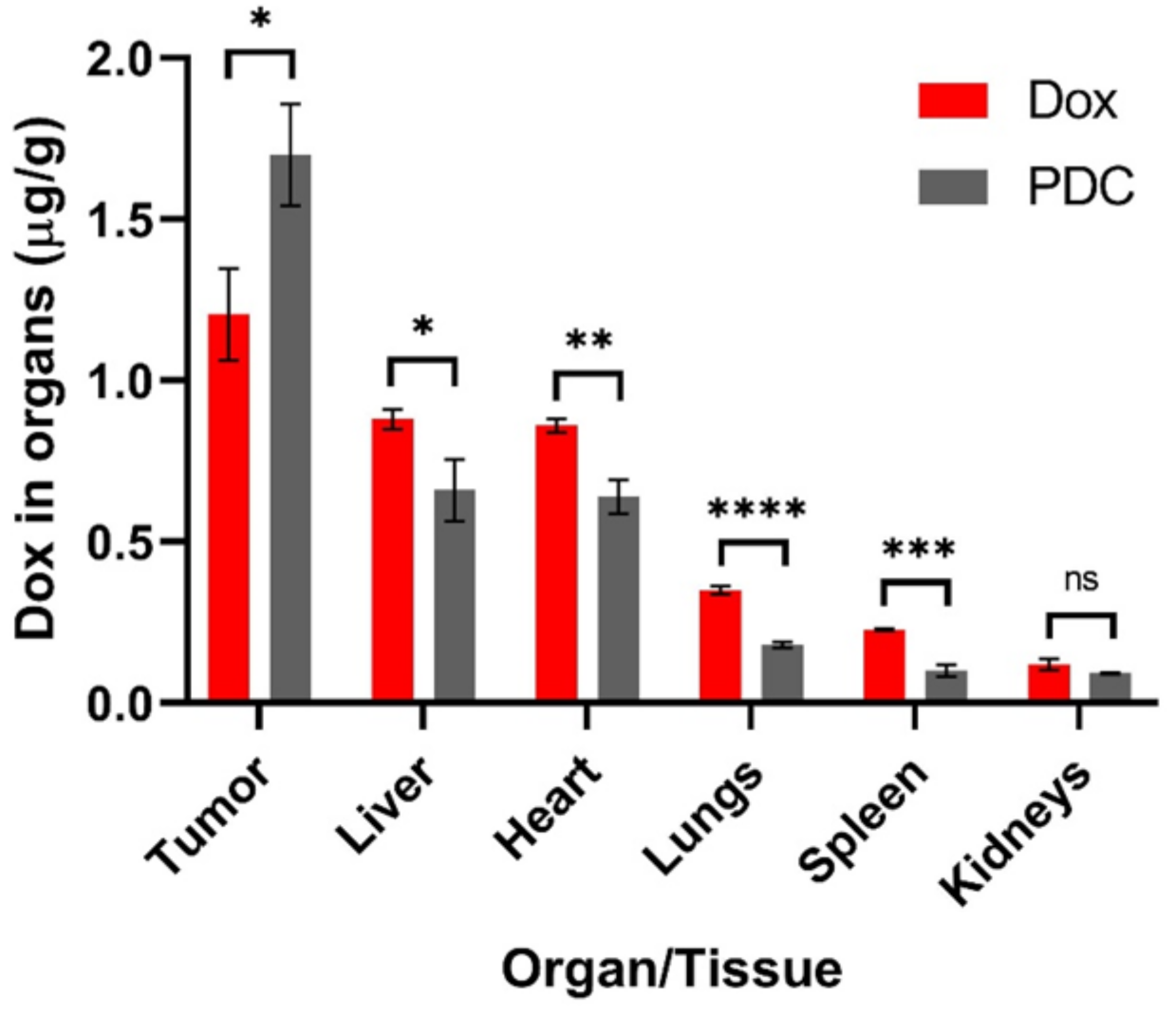
3.4. In Vivo Tissue Biodistribution Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aldox | aldoxorubicin |
| ADC ANOVA | antibody-drug conjugate analysis of variance |
| CDX | cell-derived xenograft |
| Dau | daunorubicin |
| Dox | doxorubicin |
| EphA2 | erythropoietin-producing hepatocellular receptor A2 |
| GnRH | gonadotropin-releasing hormone |
| K1 | keratin 1 |
| LC/MS | liquid chromatography-mass spectrometry |
| LRP1 | LDL receptor-related protein 1 |
| NOD-SCID | nonobese diabetic—severe combined immunodeficiency |
| PARP | poly-ADP ribose-polymerase |
| pCR | pathological complete response |
| PDC | peptide-drug conjugate or peptide-doxorubicin conjugate |
| RP-HPLC | reversed-phase high performance liquid chromatography |
| TNBC | triple-negative breast cancer |
| Trop-2 | trophoblast cell-surface antigen 2 |
References
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Palma, G.; Frasci, G.; Chirico, A.; Esposito, E.; Siani, C.; Saturnino, C.; Arra, C.; Ciliberto, G.; Giordano, A.; D’Aiuto, M. Triple negative breast cancer: Looking for the missing link between biology and treatments. Oncotarget 2015, 6, 26560–26574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Lyons, T.G. Targeted Therapies for Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 82. [Google Scholar] [CrossRef]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Mehta, R.S. Dose-dense and/or metronomic schedules of specific chemotherapies consolidate the chemosensitivity of triple-negative breast cancer: A step toward reversing triple-negative paradox. J. Clin. Oncol. 2008, 26, 3286–3288. [Google Scholar] [CrossRef] [PubMed]
- DeMichele, A.; Yee, D.; Esserman, L. Mechanisms of Resistance to Neoadjuvant Chemotherapy in Breast Cancer. N. Engl. J. Med. 2017, 377, 2287–2289. [Google Scholar] [CrossRef] [PubMed]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide-Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Mitra, A.K.; Agrahari, V.; Mandal, A.; Cholkar, K.; Natarajan, C.; Shah, S.; Joseph, M.; Trinh, H.M.; Vaishya, R.; Yang, X.; et al. Novel delivery approaches for cancer therapeutics. J. Control. Release 2015, 219, 248–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget 2015, 6, 22496–22512. [Google Scholar] [CrossRef] [Green Version]
- Sahota, S.; Vahdat, L.T. Sacituzumab govitecan: An antibody-drug conjugate. Expert Opin. Biol. Ther. 2017, 17, 1027–1031. [Google Scholar] [CrossRef]
- Ireton, R.C.; Chen, J. EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Curr. Cancer Drug Targets 2005, 5, 149–157. [Google Scholar] [CrossRef]
- Walker-Daniels, J.; Hess, A.R.; Hendrix, M.J.; Kinch, M.S. Differential regulation of EphA2 in normal and malignant cells. Am. J. Pathol. 2003, 162, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Mudd, G.E.; Brown, A.; Chen, L.; van Rietschoten, K.; Watcham, S.; Teufel, D.P.; Pavan, S.; Lani, R.; Huxley, P.; Bennett, G.S. Identification and optimization of EphA2-selective bicycles for the delivery of cytotoxic payloads. J. Med. Chem. 2020, 63, 4107–4116. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.A.; Wang, S.; Barile, E.; Das, S.K.; Emdad, L.; Sarkar, D.; De, S.K.; Morvaridi, S.K.; Stebbins, J.L.; Pandol, S.J.; et al. Therapy of pancreatic cancer via an EphA2 receptor-targeted delivery of gemcitabine. Oncotarget 2016, 7, 17103–17110. [Google Scholar] [CrossRef]
- Salem, A.F.; Wang, S.; Billet, S.; Chen, J.F.; Udompholkul, P.; Gambini, L.; Baggio, C.; Tseng, H.R.; Posadas, E.M.; Bhowmick, N.A.; et al. Reduction of Circulating Cancer Cells and Metastases in Breast-Cancer Models by a Potent EphA2-Agonistic Peptide-Drug Conjugate. J. Med. Chem. 2018, 61, 2052–2061. [Google Scholar] [CrossRef]
- Kumthekar, P.; Tang, S.C.; Brenner, A.J.; Kesari, S.; Piccioni, D.E.; Anders, C.; Carrillo, J.; Chalasani, P.; Kabos, P.; Puhalla, S.; et al. ANG1005, a Brain-Penetrating Peptide-Drug Conjugate, Shows Activity in Patients with Breast Cancer with Leptomeningeal Carcinomatosis and Recurrent Brain Metastases. Clin. Cancer Res. 2020, 26, 2789–2799. [Google Scholar] [CrossRef] [Green Version]
- Kurzrock, R.; Gabrail, N.; Chandhasin, C.; Moulder, S.; Smith, C.; Brenner, A.; Sankhala, K.; Mita, A.; Elian, K.; Bouchard, D.; et al. Safety, pharmacokinetics, and activity of GRN1005, a novel conjugate of angiopep-2, a peptide facilitating brain penetration, and paclitaxel, in patients with advanced solid tumors. Mol. Cancer Ther. 2012, 11, 308–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, C.C.; Lindenberg, M.; Bryla, C.; Patronas, N.; Peer, C.J.; Amiri-Kordestani, L.; Davarpanah, N.; Gonzalez, E.M.; Burotto, M.; Choyke, P.; et al. ANG1005 for breast cancer brain metastases: Correlation between (18)F-FLT-PET after first cycle and MRI in response assessment. Breast Cancer Res. Treat. 2016, 160, 51–59. [Google Scholar] [CrossRef]
- Chuang, N.N.; Huang, C.C. Interaction of integrin beta1 with cytokeratin 1 in neuroblastoma NMB7 cells. Biochem. Soc. Trans. 2007, 35, 1292. [Google Scholar] [CrossRef] [PubMed]
- Doljak, B.; Obermajer, N.; Jamnik, P.; Kos, J. Monoclonal antibody to cytokeratin VKIALEVEIATY sequence motif reduces plasminogen activation in breast tumour cells. Cancer Lett. 2008, 267, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Soudy, R.; Etayash, H.; Bahadorani, K.; Lavasanifar, A.; Kaur, K. Breast Cancer Targeting Peptide Binds Keratin 1: A New Molecular Marker for Targeted Drug Delivery to Breast Cancer. Mol. Pharm. 2017, 14, 593–604. [Google Scholar] [CrossRef]
- Collard, C.D.; Montalto, M.C.; Reenstra, W.R.; Buras, J.A.; Stahl, G.L. Endothelial oxidative stress activates the lectin complement pathway: Role of cytokeratin 1. Am. J. Pathol. 2001, 159, 1045–1054. [Google Scholar] [CrossRef]
- Raghuwanshi, Y.; Etayash, H.; Soudy, R.; Paiva, I.; Lavasanifar, A.; Kaur, K. Proteolytically Stable Cyclic Decapeptide for Breast Cancer Cell Targeting. J. Med. Chem. 2017, 60, 4893–4903. [Google Scholar] [CrossRef]
- Soudy, R.; Chen, C.; Kaur, K. Novel peptide-doxorubucin conjugates for targeting breast cancer cells including the multidrug resistant cells. J. Med. Chem. 2013, 56, 7564–7573. [Google Scholar] [CrossRef]
- Ziaei, E.; Saghaeidehkordi, A.; Dill, C.; Maslennikov, I.; Chen, S.; Kaur, K. Targeting Triple Negative Breast Cancer Cells with Novel Cytotoxic Peptide-Doxorubicin Conjugates. Bioconjug. Chem. 2019, 30, 3098–3106. [Google Scholar] [CrossRef]
- Banerjee, S.; Mazumdar, S. Electrospray ionization mass spectrometry: A technique to access the information beyond the molecular weight of the analyte. Int. J. Anal. Chem. 2012, 2012, 282574. [Google Scholar] [CrossRef] [Green Version]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [Green Version]
- Binkhathlan, Z.; Shayeganpour, A.; Brocks, D.R.; Lavasanifar, A. Encapsulation of P-glycoprotein inhibitors by polymeric micelles can reduce their pharmacokinetic interactions with doxorubicin. Eur. J. Pharm. Biopharm. 2012, 81, 142–148. [Google Scholar] [CrossRef]
- Shahin, M.; Soudy, R.; Aliabadi, H.M.; Kneteman, N.; Kaur, K.; Lavasanifar, A. Engineered breast tumor targeting peptide ligand modified liposomal doxorubicin and the effect of peptide density on anticancer activity. Biomaterials 2013, 34, 4089–4097. [Google Scholar] [CrossRef]
- Kai, M.; Kanaya, N.; Wu, S.V.; Mendez, C.; Nguyen, D.; Luu, T.; Chen, S. Targeting breast cancer stem cells in triple-negative breast cancer using a combination of LBH589 and salinomycin. Breast Cancer Res. Treat. 2015, 151, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Yousefpour, P.; Ahn, L.; Tewksbury, J.; Saha, S.; Costa, S.A.; Bellucci, J.J.; Li, X.; Chilkoti, A. Conjugate of Doxorubicin to Albumin-Binding Peptide Outperforms Aldoxorubicin. Small 2019, 15, e1804452. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, R.M. Patient-derived orthotopic xenografts: Better mimic of metastasis than subcutaneous xenografts. Nat. Rev. Cancer 2015, 15, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, G.L.; Sun, X.; Cao, K.X.; Ma, C.; Nan, N.; Yang, G.W.; Yu, M.W.; Wang, X.M. Establishment of a murine breast tumor model by subcutaneous or orthotopic implantation. Oncol. Lett. 2018, 15, 6233–6240. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, A.; Wang, L.; Agyin, J.; Tang, Y.; Lin, S.; Yeh, I.T.; De, K.; Sun, L.Z. Doxorubicin in combination with a small TGFbeta inhibitor: A potential novel therapy for metastatic breast cancer in mouse models. PLoS ONE 2010, 5, e10365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicki, K.T.; Sala, V.; Prever, L.; Hirsch, E.; Ardehali, H.; Ghigo, A. Preventing and Treating Anthracycline Cardiotoxicity: New Insights. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 309–332. [Google Scholar] [CrossRef]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Argenziano, M.; Gigliotti, C.L.; Clemente, N.; Boggio, E.; Ferrara, B.; Trotta, F.; Pizzimenti, S.; Barrera, G.; Boldorini, R.; Bessone, F.; et al. Improvement in the Anti-Tumor Efficacy of Doxorubicin Nanosponges in In Vitro and in Mice Bearing Breast Tumor Models. Cancers 2020, 12, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Y.; You, Y.; Chen, Y. Dual-targeting hybrid peptide-conjugated doxorubicin for drug resistance reversal in breast cancer. Int. J. Pharm. 2016, 512, 1–13. [Google Scholar] [CrossRef]
- Zhong, Q.; Bielski, E.R.; Rodrigues, L.S.; Brown, M.R.; Reineke, J.J.; da Rocha, S.R. Conjugation to Poly(amidoamine) Dendrimers and Pulmonary Delivery Reduce Cardiac Accumulation and Enhance Antitumor Activity of Doxorubicin in Lung Metastasis. Mol. Pharm. 2016, 13, 2363–2375. [Google Scholar] [CrossRef]
- Vrettos, E.I.; Mezo, G.; Tzakos, A.G. On the design principles of peptide-drug conjugates for targeted drug delivery to the malignant tumor site. Beilstein J. Org. Chem. 2018, 14, 930–954. [Google Scholar] [CrossRef]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 112–126. [Google Scholar] [CrossRef] [Green Version]
- Soudy, R.; Gill, A.; Sprules, T.; Lavasanifar, A.; Kaur, K. Proteolytically stable cancer targeting peptides with high affinity for breast cancer cells. J. Med. Chem. 2011, 54, 7523–7534. [Google Scholar] [CrossRef] [PubMed]
- Subiros-Funosas, R.; Ho, V.C.L.; Barth, N.D.; Mendive-Tapia, L.; Pappalardo, M.; Barril, X.; Ma, R.; Zhang, C.-B.; Bin-Zhi, Q.; Miquel, S.; et al. Fluorogenic Trp(redBODIPY) cyclopeptide targeting keratin 1 for imaging of aggressive carcinomas. Chem. Sci. 2020, 11, 1368. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, G.; Lanza, M.; Kratz, F.; Merina, L.; Fiume, L. A novel method for coupling doxorubicin to lactosaminated human albumin by an acid sensitive hydrazone bond: Synthesis, characterization and preliminary biological properties of the conjugate. Eur. J. Pharm. Sci. 2004, 23, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F. DOXO-EMCH (INNO-206): The first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin. Investig. Drugs 2007, 16, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F.; Warnecke, A.; Schmid, B.; Chung, D.E.; Gitzel, M. Prodrugs of anthracyclines in cancer chemotherapy. Curr. Med. Chem. 2006, 13, 477–523. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.P.; Papai, Z.; Mukhametshina, G.; Sankhala, K.; Vasylyev, L.; Fedenko, A.; Khamly, K.; Ganjoo, K.; Nagarkar, R.; Wieland, S.; et al. First-line aldoxorubicin vs doxorubicin in metastatic or locally advanced unresectable soft-tissue sarcoma: A phase 2b randomized clinical trial. JAMA Oncol. 2015, 1, 1272–1280. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Yan, J.; Forscher, C.; Hendifar, A. Aldoxorubicin: A tumor-targeted doxorubicin conjugate for relapsed or refractory soft tissue sarcomas. Drug Des. Dev. Ther. 2018, 12, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Randelovic, I.; Schuster, S.; Kapuvari, B.; Fossati, G.; Steinkuhler, C.; Mezo, G.; Tovari, J. Improved in vivo anti-tumor and anti-metastatic effect of GnRH-III-daunorubicin analogs on colorectal and breast carcinoma bearing mice. Int. J. Mol. Sci. 2019, 20, 4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditzel, H.J.; Garrigues, U.; Andersen, C.B.; Larsen, M.K.; Garrigues, H.J.; Svejgaard, A.; Hellstrom, I.; Hellstrom, K.E.; Jensenius, J.C. Modified cytokeratins expressed on the surface of carcinoma cells undergo endocytosis upon binding of human monoclonal antibody and its recombinant Fab fragment. Proc. Natl. Acad. Sci. USA 1997, 94, 8110–8115. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saghaeidehkordi, A.; Chen, S.; Yang, S.; Kaur, K. Evaluation of a Keratin 1 Targeting Peptide-Doxorubicin Conjugate in a Mouse Model of Triple-Negative Breast Cancer. Pharmaceutics 2021, 13, 661. https://doi.org/10.3390/pharmaceutics13050661
Saghaeidehkordi A, Chen S, Yang S, Kaur K. Evaluation of a Keratin 1 Targeting Peptide-Doxorubicin Conjugate in a Mouse Model of Triple-Negative Breast Cancer. Pharmaceutics. 2021; 13(5):661. https://doi.org/10.3390/pharmaceutics13050661
Chicago/Turabian StyleSaghaeidehkordi, Azam, Shiuan Chen, Sun Yang, and Kamaljit Kaur. 2021. "Evaluation of a Keratin 1 Targeting Peptide-Doxorubicin Conjugate in a Mouse Model of Triple-Negative Breast Cancer" Pharmaceutics 13, no. 5: 661. https://doi.org/10.3390/pharmaceutics13050661
APA StyleSaghaeidehkordi, A., Chen, S., Yang, S., & Kaur, K. (2021). Evaluation of a Keratin 1 Targeting Peptide-Doxorubicin Conjugate in a Mouse Model of Triple-Negative Breast Cancer. Pharmaceutics, 13(5), 661. https://doi.org/10.3390/pharmaceutics13050661