
Cyclodextrin Inclusion Complexes of Auranofin and Its Iodido Analog: A Chemical and Biological Study

 ,
, 

 and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Analytical Methods for Drug Determination
2.3. Phase Solubility Studies
2.4. NMR Studies
2.5. Binary Systems Preparation Methods
2.6. Differential Scanning Calorimetry (DSC)
2.7. Powder X-Ray Diffractometry (PXRD)
2.8. Cell Lines and Culture Conditions
2.9. Cytotoxicity Assays
3. Results and Discussion
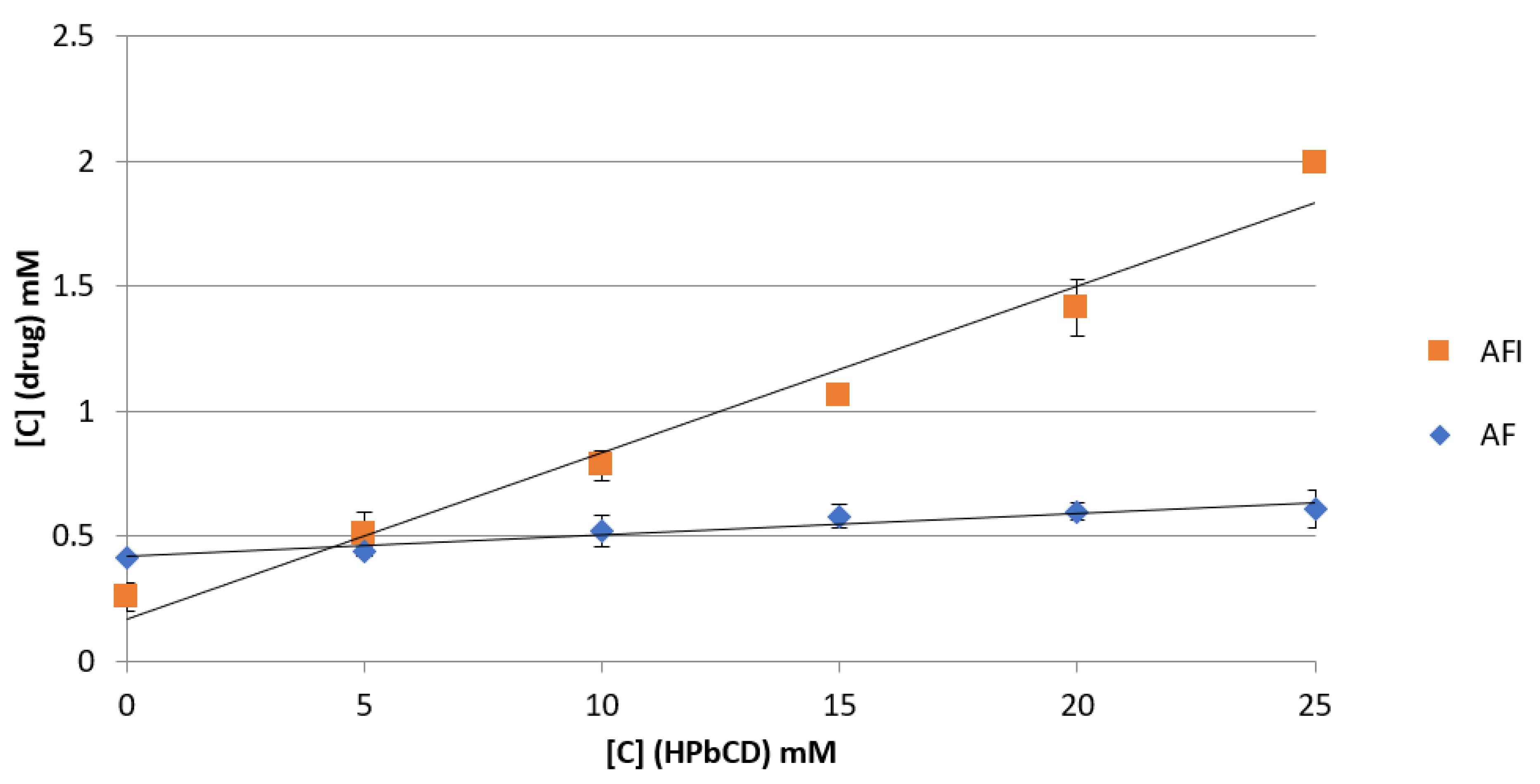
3.1. Phase Solubility Studies
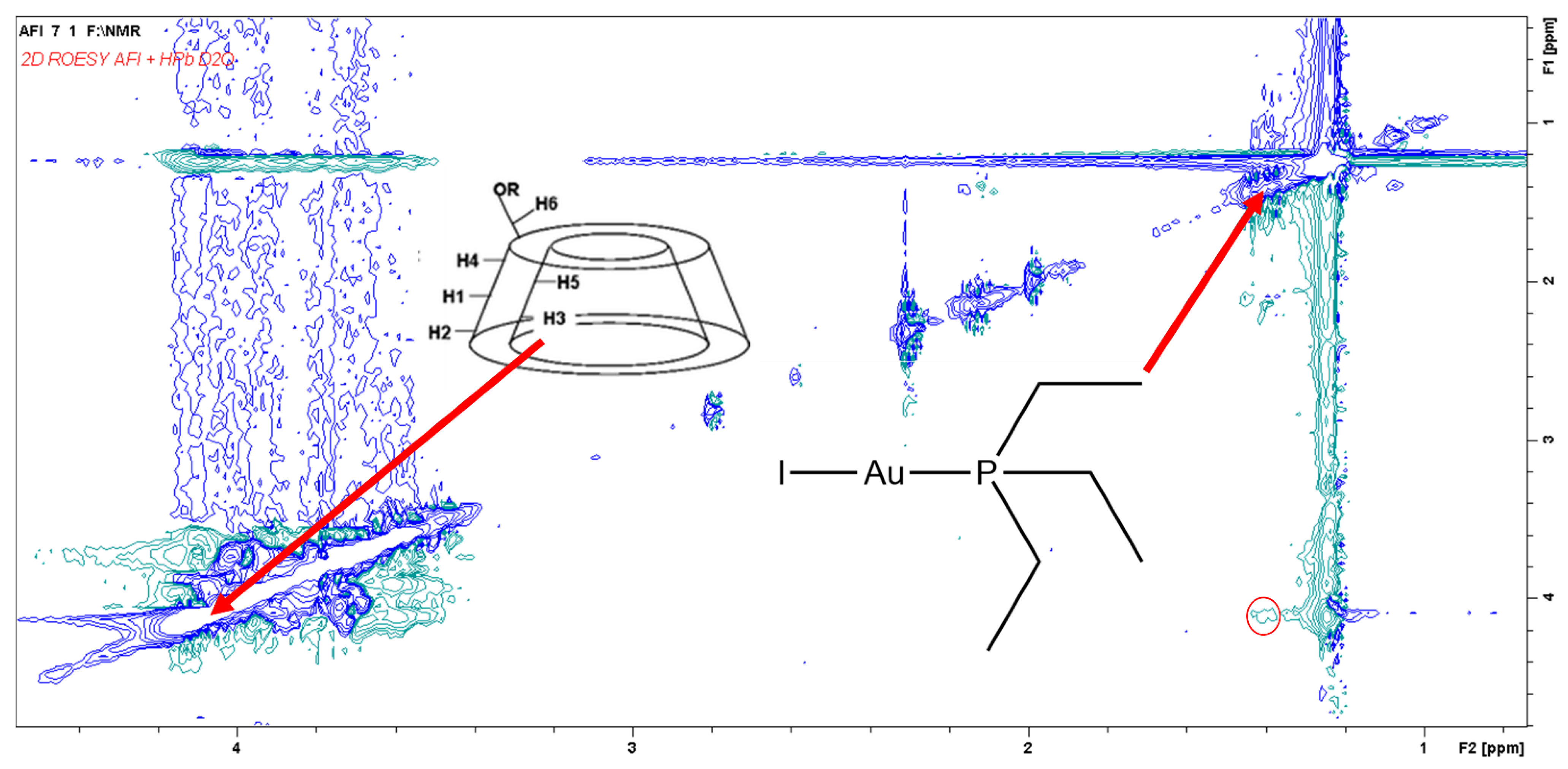
3.2. NMR Studies on the AFI/HPβ–CD System
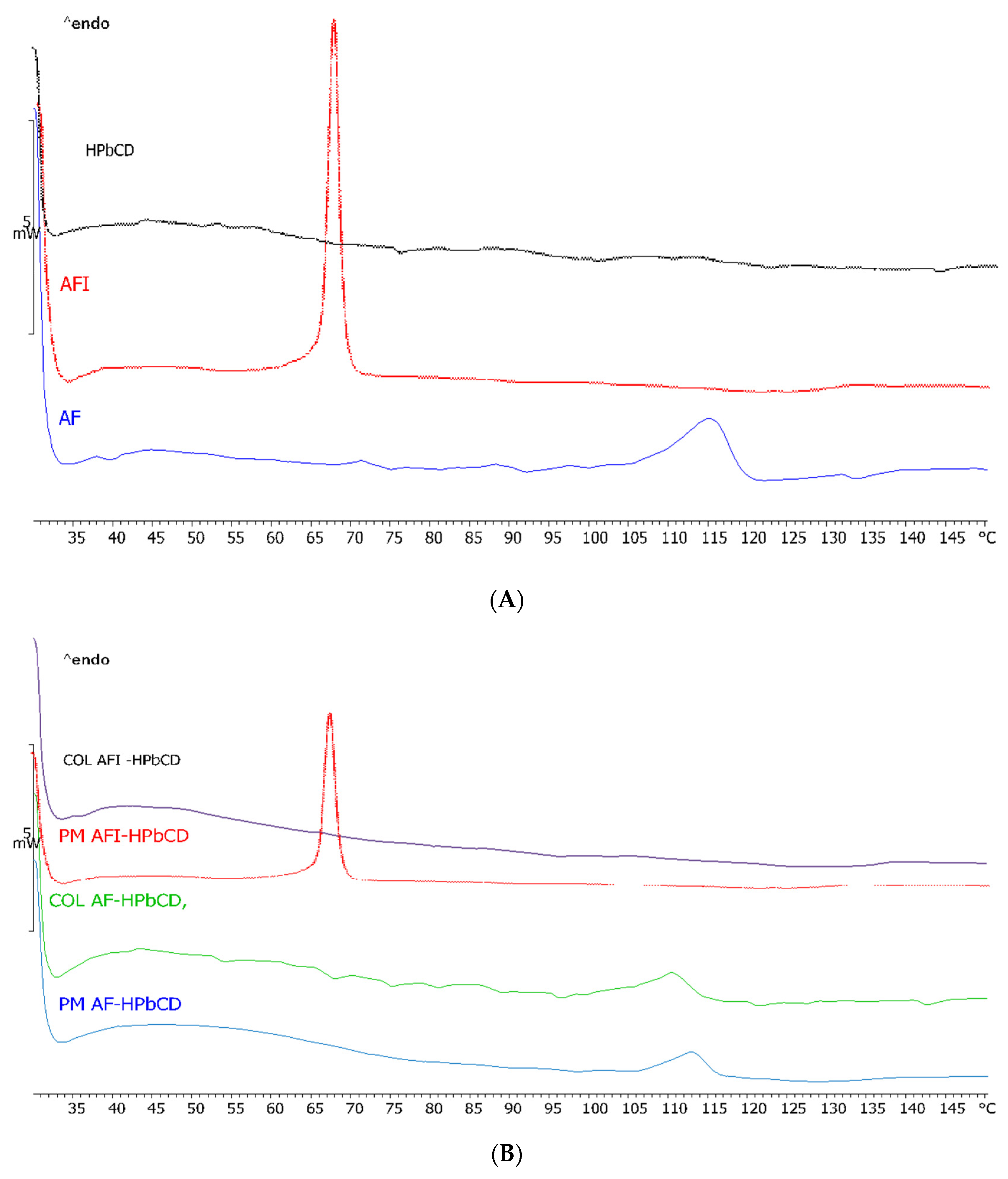
3.3. Binary System Characterization in the Solid State
3.4. Biological Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ClinicalTrials. Available online: https://www.clinicaltrials.gov (accessed on 11 January 2021).
- Marzo, T.; Cirri, D.; Gabbiani, C.; Gamberi, T.; Magherini, F.; Pratesi, A.; Guerri, A.; Biver, T.; Binacchi, F.; Stefanini, M.; et al. Auranofin, Et3PAuCl, and Et3PAuI Are Highly Cytotoxic on Colorectal Cancer Cells: A Chemical and Biological Study. ACS Med. Chem. Lett. 2017, 8, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Marzo, T.; Cirri, D.; Pollini, S.; Prato, M.; Fallani, S.; Cassetta, M.I.; Novelli, A.; Rossolini, G.M.; Messori, L. Auranofin and its Analogues Show Potent Antimicrobial Activity against Multidrug-Resistant Pathogens: Structure–Activity Relationships. ChemMedChem 2018, 13, 2448–2454. [Google Scholar] [CrossRef]
- Marzo, T.; Massai, L.; Pratesi, A.; Stefanini, M.; Cirri, D.; Magherini, F.; Becatti, M.; Landini, I.; Nobili, S.; Mini, E.; et al. Replacement of the Thiosugar of Auranofin with Iodide Enhances the Anticancer Potency in a Mouse Model of Ovarian Cancer. ACS Med. Chem. Lett. 2019, 10, 656–660. [Google Scholar] [CrossRef]
- Blodgett, R.C., Jr. Auranofin: Experience to date. Am. J. Med. 1983, 75, 86–89. [Google Scholar] [CrossRef]
- Tomasello, M.F.; Nardon, C.; Lanza, V.; Di Natale, G.; Pettenuzzo, N.; Salmaso, S.; Milardi, D.; Caliceti, P.; Pappalardo, G.; Fregona, D. New comprehensive studies of a gold(III) Dithiocarbamate complex with proven anticancer properties: Aqueous dissolution with cyclodextrins, pharmacokinetics and upstream inhibition of the ubiquitin-proteasome pathway. Eur. J. Med. Chem. 2017, 138, 115–127. [Google Scholar] [CrossRef]
- Wani, W.A.; Prashar, S.; Shreaz, S.; Gómez-Ruiz, S. Nanostructured materials functionalized with metal complexes: In search of alternatives for administering anticancer metallodrugs. Coord. Chem. Rev. 2016, 312, 67–98. [Google Scholar] [CrossRef]
- Duchêne, D. New Trends in Cyclodextrins and Derivatives; Editions de Santé: Paris, France, 1991. [Google Scholar]
- Sharma, N.; Baldi, A. Exploring versatile applications of cyclodextrins: An overview. Drug Deliv. 2014, 23, 729–747. [Google Scholar] [CrossRef] [PubMed]
- Inactive Ingredient Search for Approved Drug Products. Available online: http://www.accessdata.fda.gov/scripts/cder/iig/getiigWEB.cfm (accessed on 4 April 2020).
- Higuchi, T.; Connors, K.A. Phase-Solubility Techniques. In Advances in Analytical Chemistry and Instrumentation; Reilly, C.N., Ed.; Wiley-Intersciences: New York, NY, USA, 1965; Volume 4, pp. 117–212. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-M.; Yu, S.-C.; Tsai, F.-J.; Tsai, Y. Enhancement of rhubarb extract solubility and bioactivity by 2-hydroxypropyl-β-cyclodextrin. Carbohydr. Polym. 2013, 98, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Kerč, J.; Srcic, S.; Mohar, M.; Kofler, B. Preparation and evaluation of auranofin solvent deposits onto lactose. Acta Pharm. 1996, 46, 125–130. [Google Scholar]
- Mura, P.; Corti, G.; Cirri, M.; Maestrelli, F.; Mennini, N.; Bragagni, M. Development of mucoadhesive films for buccal administration of flufenamic acid: Effect of cyclodextrin complexation. J. Pharm. Sci. 2010, 99, 3019–3029. [Google Scholar] [CrossRef] [PubMed]
- Landini, I.; Massai, L.; Cirri, D.; Gamberi, T.; Paoli, P.; Messori, L.; Mini, E.; Nobili, S. Structure-activity relationships in a series of auranofin analogues showing remarkable antiproliferative properties. J. Inorg. Biochem. 2020, 208, 111079. [Google Scholar] [CrossRef] [PubMed]
- Talib, J.; Beck, J.L.; Ralph, S.F. A mass spectrometric investigation of the binding of gold antiarthritic agents and the metabolite [Au(CN)2]− to human serum albumin. J. Biol. Inorg. Chem. 2006, 11, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Coffer, M.T.; Shaw, C.F., III; Eidsness, M.K.; Watkins, J.W., II; Elder, R.C. Reactions of auranofin and chloro(triethylphosphine)gold with bovine serum albumin. Inorg. Chem. 1986, 25, 333–339. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 Mean ± SD (μM) | ||||
|---|---|---|---|---|
| Auranofin | Auranofin/HPβ–CD | AFI | AFI/HPβ–CD | |
| A2780 | 0.611 ± 0.085 | 0.831 ± 0.079 | 0.898 ± 0.117 | 0.910 ± 0.162 |
| SKOV3 | 0.360 ± 0.004 | 0.577 ± 0.177 | 0.685 ± 0.100 | 0.535 ± 0.188 |
| IGROV-1 | 0.657 ± 0.164 | 1.092 ± 0.045 | 0.856 ± 0.018 | 0.684 ± 0.158 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cirri, D.; Landini, I.; Massai, L.; Mini, E.; Maestrelli, F.; Messori, L. Cyclodextrin Inclusion Complexes of Auranofin and Its Iodido Analog: A Chemical and Biological Study. Pharmaceutics 2021, 13, 727. https://doi.org/10.3390/pharmaceutics13050727
Cirri D, Landini I, Massai L, Mini E, Maestrelli F, Messori L. Cyclodextrin Inclusion Complexes of Auranofin and Its Iodido Analog: A Chemical and Biological Study. Pharmaceutics. 2021; 13(5):727. https://doi.org/10.3390/pharmaceutics13050727
Chicago/Turabian StyleCirri, Damiano, Ida Landini, Lara Massai, Enrico Mini, Francesca Maestrelli, and Luigi Messori. 2021. "Cyclodextrin Inclusion Complexes of Auranofin and Its Iodido Analog: A Chemical and Biological Study" Pharmaceutics 13, no. 5: 727. https://doi.org/10.3390/pharmaceutics13050727
APA StyleCirri, D., Landini, I., Massai, L., Mini, E., Maestrelli, F., & Messori, L. (2021). Cyclodextrin Inclusion Complexes of Auranofin and Its Iodido Analog: A Chemical and Biological Study. Pharmaceutics, 13(5), 727. https://doi.org/10.3390/pharmaceutics13050727