
How Do Hospital Pharmacists Approach Substitution of Nanomedicines? Insights from a Qualitative Pilot Study and a Quantitative Market Research Analysis in Five European Countries

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Qualitative Pilot Study
2.2. Quantitative Market Research
3. Results
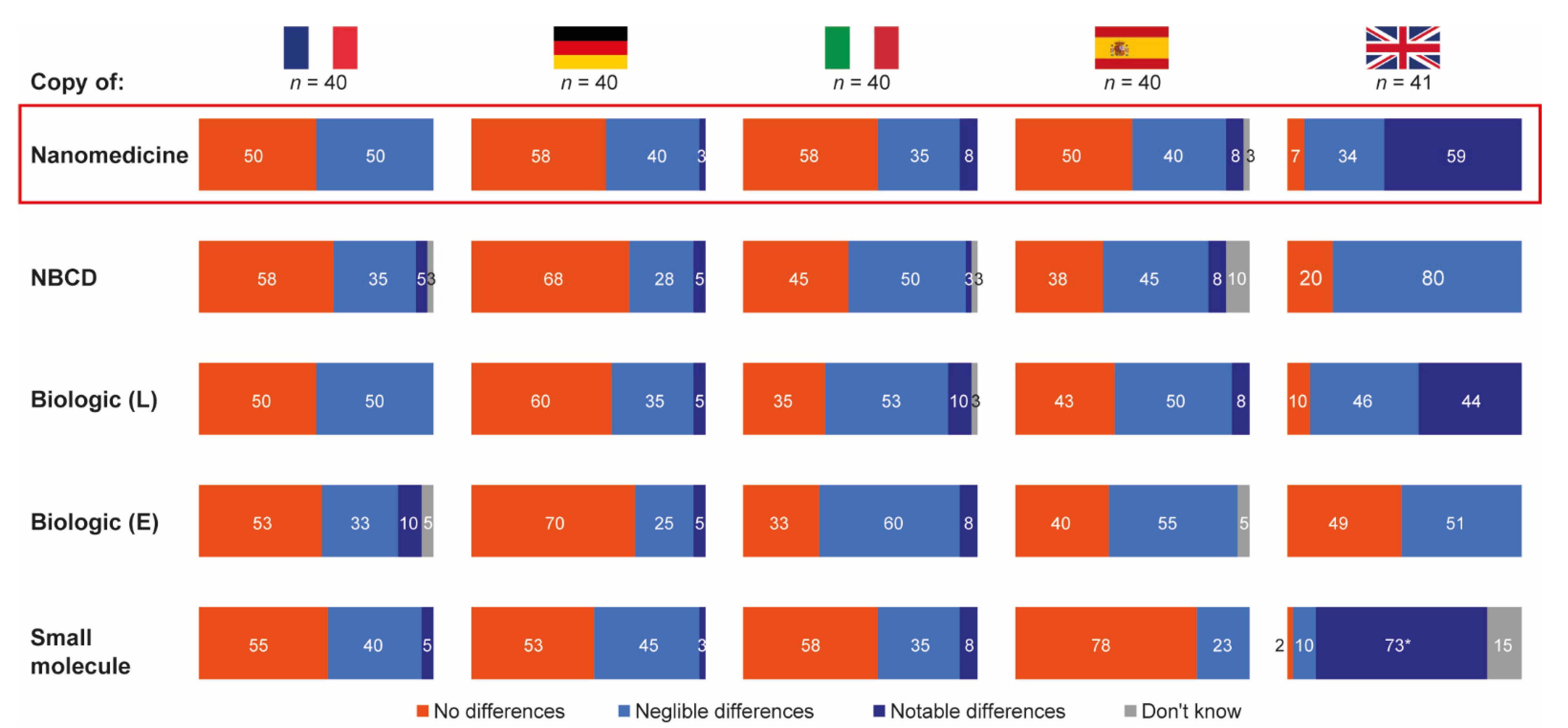
3.1. Perception of Drug Substitution and Use of Follow-On Products
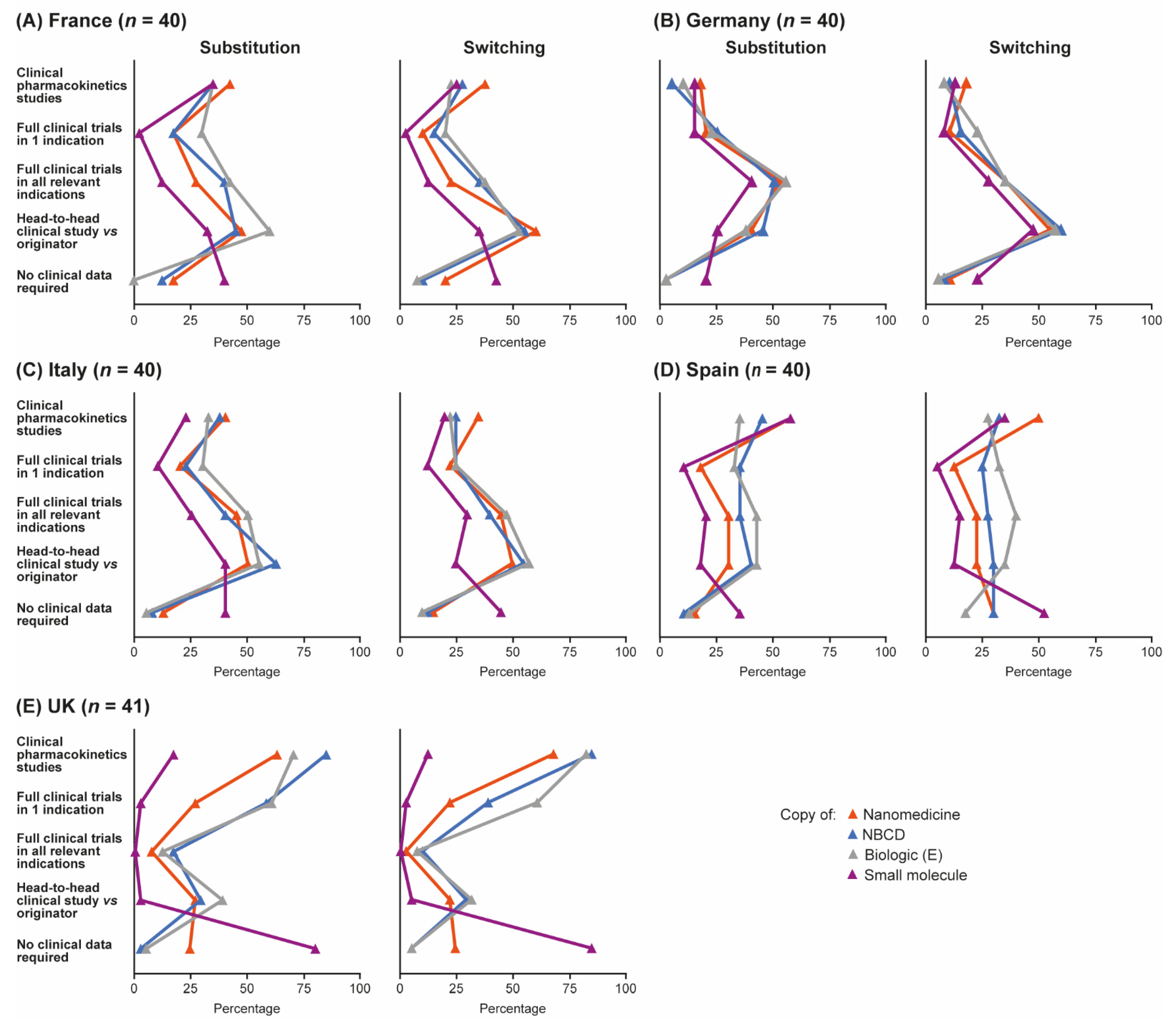
3.2. Data Requirements for Substitution in the Hospital Formulary
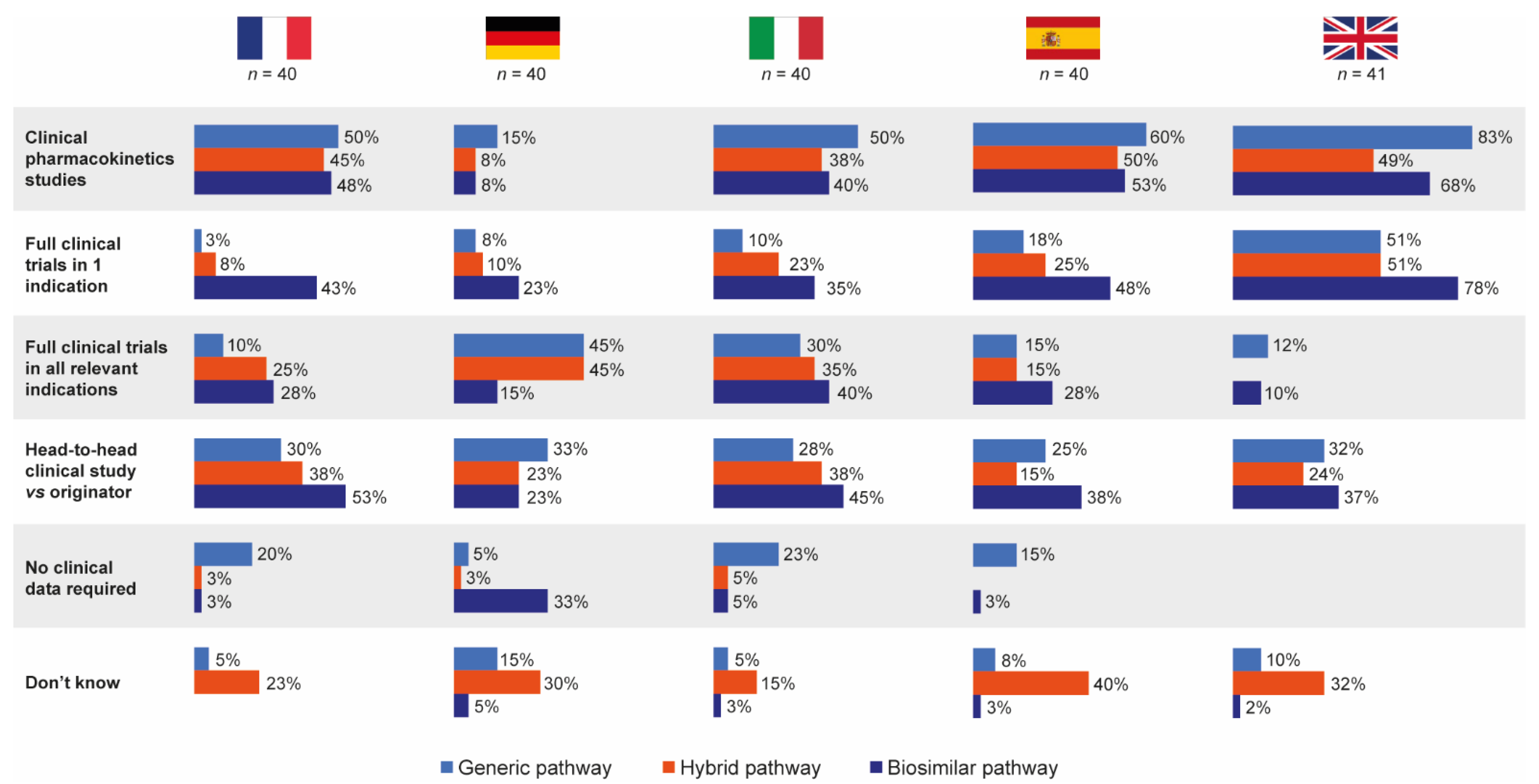
3.3. Knowledge of Data Requirements for Follow-On Products
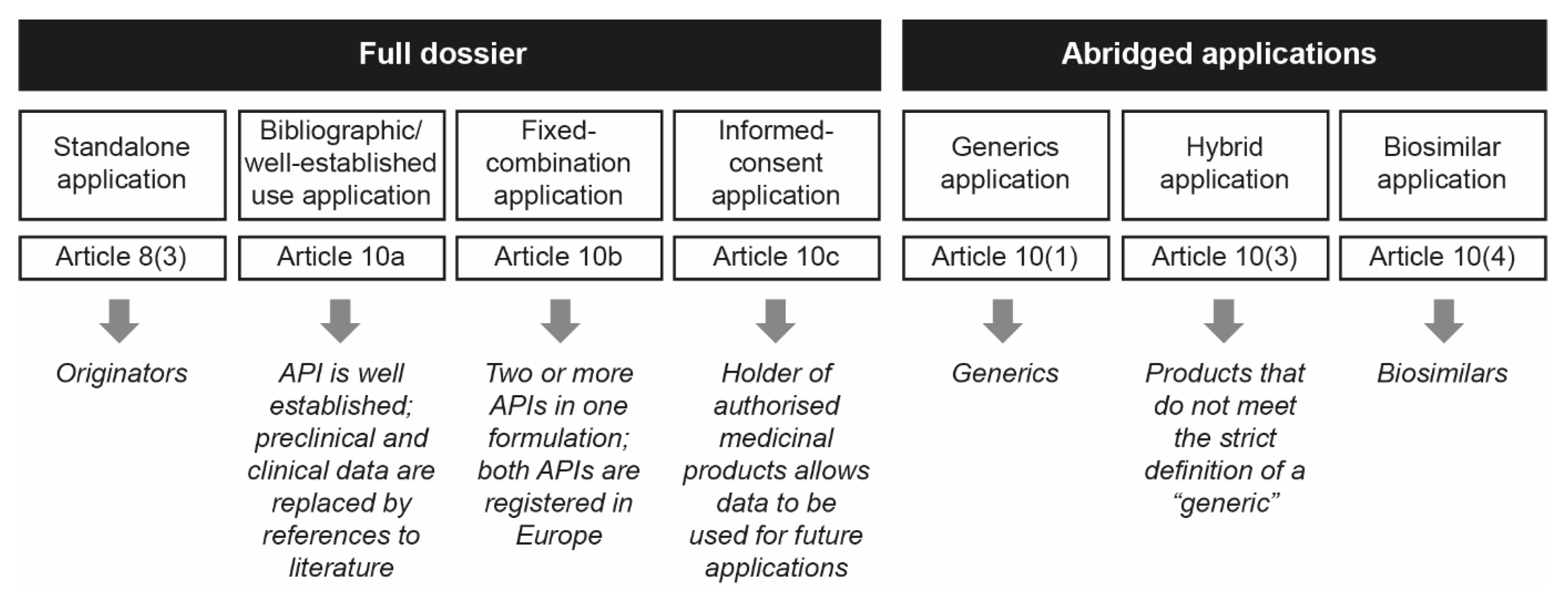
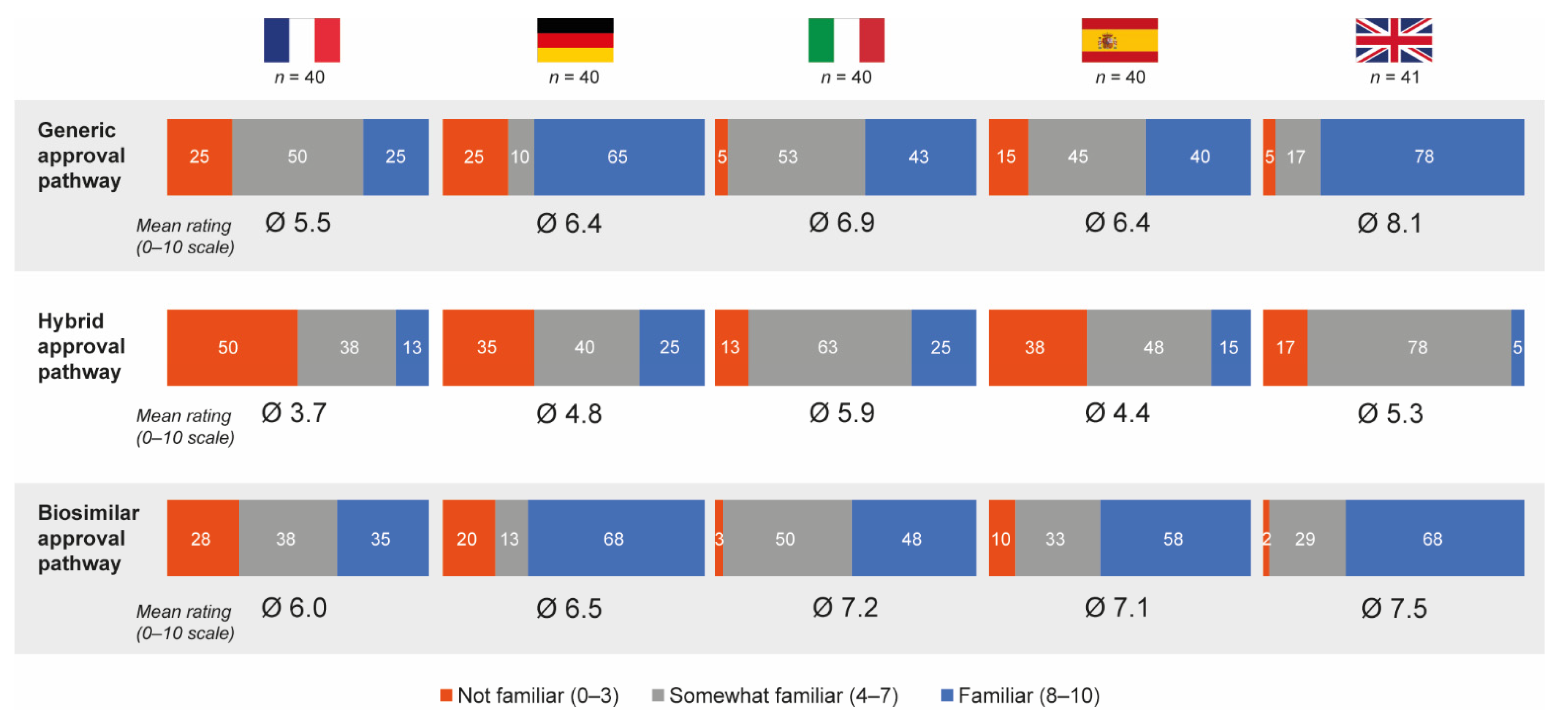
3.4. Awareness of European Regulatory Approval Pathways
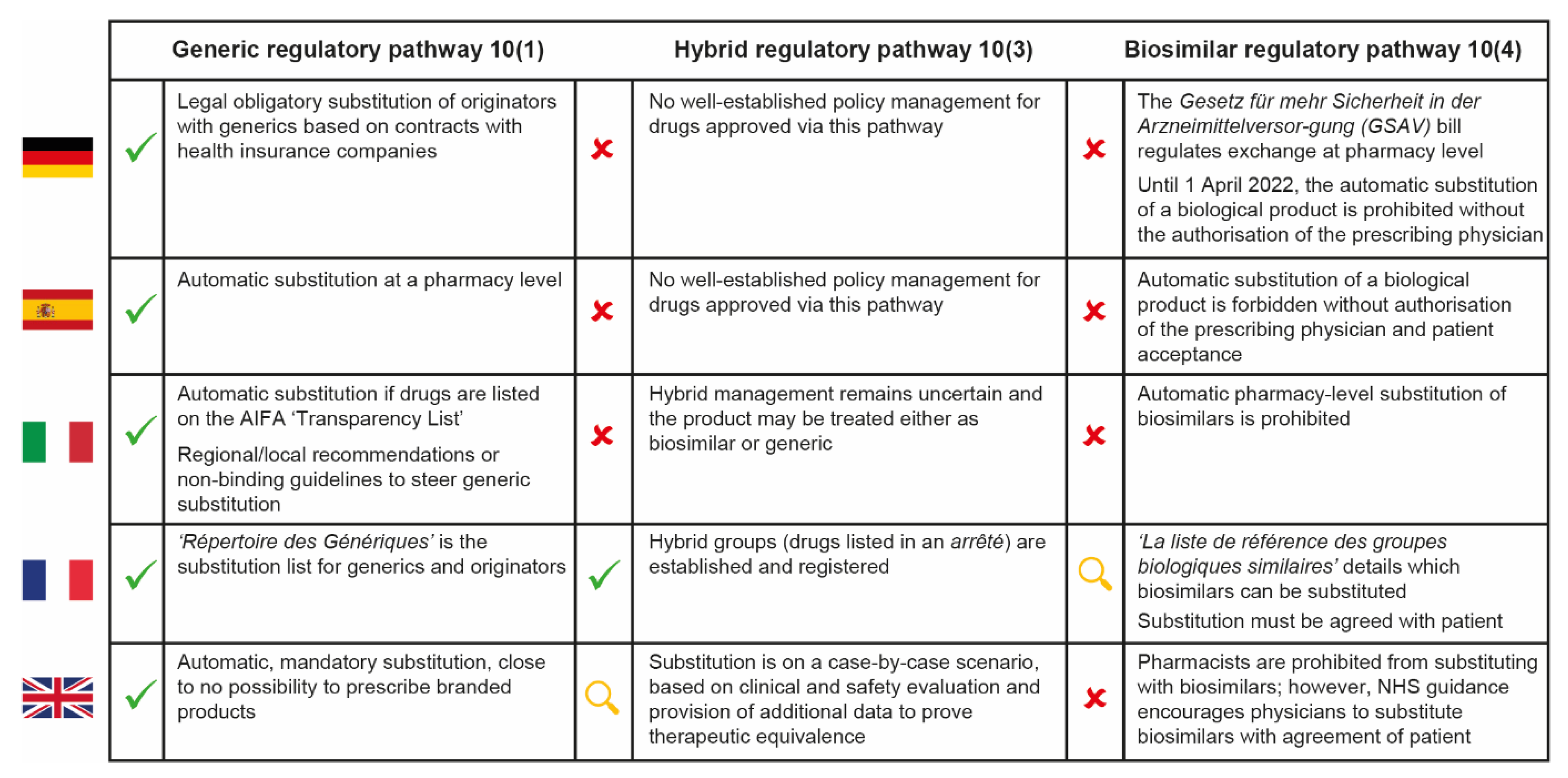
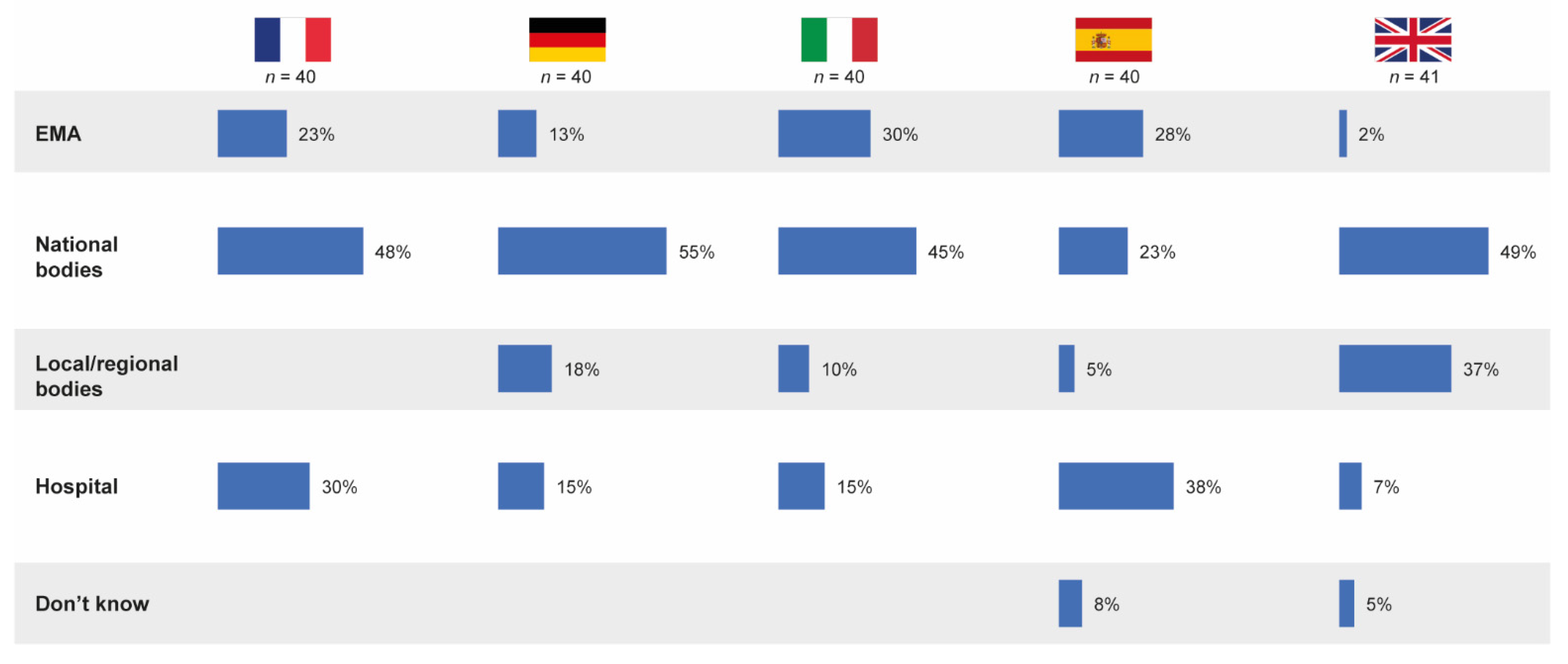
3.5. Responsibility for Defining Substitutability
3.6. Inclusion of Hybrid Pathway-Approved Follow-Ons in the Hospital Formulary: Who Decides?
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soares, S.; Sousa, J.; Pais, A.; Vitorino, C. Nanomedicine: Principles, properties, and regulatory issues. Front. Chem. 2018, 6, 360. [Google Scholar] [CrossRef] [PubMed]
- Astier, A.; Barton Pai, A.; Bissig, M.; Crommelin, D.J.A.; Flühmann, B.; Hecq, J.D.; Knoeff, J.; Lipp, H.P.; Morell-Baladrón, A.; Mühlebach, S. How to select a nanosimilar. Ann. N. Y. Acad. Sci. 2017, 1407, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Rocco, P.; Musazzi, U.M.; Franzè, S.; Minghetti, P. Copies of nonbiological complex drugs: Generic, hybrid or biosimilar? Drug Discov. Today 2019, 24, 250–255. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.R.; Cruz, C.N.; Chen, M.L.; Kapoor, M.; Lee, S.L.; Tyner, K.M. The evolving landscape of drug products containing nanomaterials in the United States. Nat. Nanotechnol. 2017, 12, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Roco, M.; Müller, B.; Wagner, E.; Borchard, G.; Francesco, T.D.; Jurczyk, K.; Braegger, U.; Jurczyk, M.; Bartolucci, C.; Ijabadeniyi, O.; et al. Nanoscience and Nanotechnology. Advances and Developments in Nano-Sized Materials; Van de Voorde, M., Ed.; De Gruyter: Berlin, Germany, 2018. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, R.S.; Silva-Lima, B.; Magro, F.; Alcobia, A.; da Costa, F.L.; Feio, J. Non-biological complex drugs (NBCDs): Complex pharmaceuticals in need of individual robust clinical assessment before any therapeutic equivalence decision. Front. Med. 2020, 7, 590527. [Google Scholar] [CrossRef] [PubMed]
- Mühlebach, S. Regulatory challenges of nanomedicines and their follow-on versions: A generic or similar approach? Adv. Drug Deliv. Rev. 2018, 131, 122–131. [Google Scholar] [CrossRef]
- Musazzi, U.M.; Marini, V.; Casiraghi, A.; Minghetti, P. Is the European regulatory framework sufficient to assure the safety of citizens using health products containing nanomaterials? Drug Discov. Today 2017, 22, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Pita, R.; Ehmann, F.; Papaluca, M. Nanomedicines in the EU–Regulatory Overview. AAPS J. 2016, 18, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Stolk, P.; De Bruin, M.L.; Leufkens, H.G.M.; Crommelin, D.J.A.; De Vlieger, J.S.B. The EU regulatory landscape of non-biological complex drugs (NBCDs) follow-on products: Observations and recommendations. Eur. J. Pharm. Sci. 2019, 133, 228–235. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. EMEA/CHMP/225411/2006. European Medicines Agency Procedural Advice for Users of the Centralised Procedure for Generic/Hybrid Applications. Available online: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/european-medicines-agency-procedural-advice-users-centralised-procedure-generic/hybrid-applications_en.pdf (accessed on 28 April 2021).
- Hussaarts, L.; Muhlebach, S.; Shah, V.P.; McNeil, S.; Borchard, G.; Fluhmann, B.; Weinstein, V.; Neervannan, S.; Griffiths, E.; Jiang, W.; et al. Equivalence of complex drug products: Advances in and challenges for current regulatory frameworks. Ann. N. Y. Acad. Sci. 2017, 1407, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Legal Basis & Types of Approvals. Available online: https://www.ema.europa.eu/en/documents/presentation/presentation-legal-basis-types-approvals-s-prilla_en.pdf (accessed on 28 April 2021).
- Minghetti, P.; Musazzi, U.M.; Casiraghi, A.; Rocco, P. Old active ingredients in new medicinal products: Is the regulatory path coherent with patients’ expectations? Drug Discov. Today 2020, 25, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Flühmann, B.; Ntai, I.; Borchard, G.; Simoens, S.; Mühlebach, S. Nanomedicines: The magic bullets reaching their target? Eur. J. Pharm. Sci. 2019, 128, 73–80. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Reflection Paper on the Data Requirements for Intravenous Iron-Based Nano-Colloidal Products Developed with Reference to an Innovator Medicinal Product. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-data-requirements-intravenous-iron-based-nano-colloidal-products-developed_en.pdf (accessed on 28 April 2021).
- European Medicines Agency; European Commission. Biosimilars in the EU. Information for Healthcare Professionals. Available online: https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf (accessed on 28 April 2021).
- Michalopoulos, S.; on Behalf of EURACTIV. MEP: Parliament Will Pressure Commission to Focus on Nanomedicines. Available online: https://www.euractiv.com/section/health-consumers/news/mep-parliament-will-pressure-commission-to-focus-on-nanomedicines/ (accessed on 28 April 2021).
- Ehmann, F.; Pita, R. The EU is ready for non-biological complex medicinal products. Generics Biosimilars Initiat. J. 2016, 5, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Gradl, G.; Krieg, E.M.; Schulz, M. Evaluation of pharmaceutical concerns in Germany: Frequency and potential reasons. Pharm. Pract. 2016, 14, 786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondelo-Garcia, C.; Mendoza, E.; Movilla-Fernandez, M.J.; Coronado, C. Perceptions of pharmacists and physicians on generic substitution in a financial crisis context in Northwestern Spain: A qualitative study. Health Policy 2018, 122, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Duerden, M.G.; Hughes, D.A. Generic and therapeutic substitutions in the UK: Are they a good thing? Br. J. Clin. Pharmacol. 2010, 70, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.; Asmar, R.; Dahlof, B.; Hill, K.; Jones, D.A.; Jordan, J.; Livingston, M.; Macgregor, G.; Sobanja, M.; Stafylas, P.; et al. Generic and therapeutic substitution: A viewpoint on achieving best practice in Europe. Br. J. Clin. Pharmacol. 2011, 72, 727–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Légifrance. Order of 12 November 2019 Specifying, Under Article L. 5125-23 of the Public Health Code, Medical Situations in Which the Substitution of a Specialty of the Same Generic Group May Be Excluded. Available online: https://www.legifrance.gouv.fr/jorf/id/JORFTEXT000039393124/ (accessed on 28 April 2021).
- Hertig, J.; Shah, V.P.; Flühmann, B.; Mühlebach, S.; Stemer, G.; Surugue, J.; Moss, R.; Di Francesco, T. Tackling the challenges of nanomedicines: Are we ready? Am. J. Health Syst. Pharm. 2021, 78, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Italian Medicines Agency (AIFA). Transparency Lists. Available online: https://www.aifa.gov.it/en/liste-di-trasparenza (accessed on 28 April 2021).
- Moorkens, E.; Vulto, A.G.; Huys, I.; Dylst, P.; Godman, B.; Keuerleber, S.; Claus, B.; Dimitrova, M.; Perova, G.; Sovic-Brkicic, L.; et al. Policies for biosimilar uptake in Europe: An overview. PLoS ONE 2017, 12, e0190147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italian Medicines Agency (AIFA). Biosimilar Medicines. Available online: https://www.aifa.gov.it/en/farmaci-biosimilari (accessed on 28 April 2021).
- Delgado, M. Biosimilar Automatic Substitution in the EU5: Current State & Future Outlook. Available online: https://www.biosimilardevelopment.com/doc/biosimilar-automatic-substitution-in-the-eu-current-state-future-outlook-0001#:~:text=In%20Spain%2C%20biosimilars%20are%20not,prescribing%20physician%20and%20patient%20acceptance (accessed on 28 April 2021).
- Agence Nationale de Sécurité du Medicament et des Produits de Santé (ANSM). Décision du 12 JAN. 2021. Portant Modification au Répertoire des Groupes Génériques Mentionné à l’Article R. 5121-5 du CODE de la Santé Publique. Available online: https://ansm.sante.fr/Decisions/Generiques-biosimilaires-medicaments-en-acces-direct-depot-de-publicite-PSL-MDS-bonnes-pratiques-Repertoire-des-generiques (accessed on 28 April 2021).
- Agence Nationale de Sécurité du Medicament et des Produits de Santé (ANSM). Les Médicaments Biosimilaires. Available online: https://www.ansm.sante.fr/Activites/Medicaments-biosimilaires/Les-medicaments-biosimilaires/(offset)/0 (accessed on 28 April 2021).
- National Health Service (NHS) England. Commissioning Framework for Biological Medicines (Including Biosimilar Medicines). Available online: https://www.england.nhs.uk/wp-content/uploads/2017/09/biosimilar-medicines-commissioning-framework.pdf (accessed on 28 April 2021).
- Federal Ministry of Health (Germany). Law for More Safety in the Supply of Medicines (GSAV). Available online: https://www.bundesgesundheitsministerium.de/gsav.html#:~:text=Das%20ist%20Ziel%20des%20Gesetzes,sowie%20Herstellbetrieben%20st%C3%A4rker%20kontrolliert%20werden (accessed on 28 April 2021).
- Steinmeier, F.-W.; Merkel, A.; Spahn, J. Gesetz für mehr Sicherheit in der Arzneimittelversorgung. Das Bundesgesetzblatt 2019, 30, 1202–1220. [Google Scholar]
- PHARMADOC. Intercambiabilitá Copemyl/Copaxone: Il Consiglio di Stato dà Ragione ad AIFA. Available online: https://www.pharmadoc.it/intercambiabilita-copemyl-copaxone-il-consiglio-di-stato-da-ragione-ad-aifa/ (accessed on 28 April 2021).
- International Pharmaceutical Federation (FIP). FIP Statement of Policy—Pharmacist’s Authority in Pharmaceutical Product Selection: Therapeutic Interchange and Substitution. 2018. Available online: https://www.fip.org/file/2086 (accessed on 28 April 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of institution | Hospital/clinic |
| Role in the institution | Pharmacist, pharmacist purchasing manager |
| Years of experience | 3–30 |
| Age | 30–60 years |
| Extent of involvement in procurement and listing of off-patent pharmaceuticals | Be sole decision-maker for purchasing and listing Be one of the main decision-makers for purchasing and listing Have some influence on purchasing and listing, but not a main decision-maker |
| Extent of involvement in setting hospital guidelines and protocols | Be solely responsible for setting guidelines and protocols for drug substitution Be one of the main decision-makers for setting guidelines and protocols for drug substitution Have some influence on setting guidelines and protocols for drug substitution, but not a main decision-maker |
| Type of institution, n (%) | |
| 201 (100) |
| Role in the institution, n (%) | |
| 88 (44) 11 (5) 83 (41) 19 (9) |
| Mean (standard deviation) years of experience | 17.4 (6.4) |
| Mean (standard deviation) age, years | 45.6 (6.9) |
| Extent of involvement in procurement and listing of off-patent pharmaceuticals, n (%) | |
| 37 (18) 142 (71) 22 (11) |
| Extent of involvement in setting hospital guidelines and protocols for drug substitution, n (%) | |
| 23 (11) 156 (78) 22 (11) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sofia, N.; Mühlebach, S.; Musazzi, U.M.; Khatib, R.; Martinez Sesmero, J.M.; Lipp, H.-P.; Surugue, J.; Di Francesco, T.; Flühmann, B. How Do Hospital Pharmacists Approach Substitution of Nanomedicines? Insights from a Qualitative Pilot Study and a Quantitative Market Research Analysis in Five European Countries. Pharmaceutics 2021, 13, 1010. https://doi.org/10.3390/pharmaceutics13071010
Sofia N, Mühlebach S, Musazzi UM, Khatib R, Martinez Sesmero JM, Lipp H-P, Surugue J, Di Francesco T, Flühmann B. How Do Hospital Pharmacists Approach Substitution of Nanomedicines? Insights from a Qualitative Pilot Study and a Quantitative Market Research Analysis in Five European Countries. Pharmaceutics. 2021; 13(7):1010. https://doi.org/10.3390/pharmaceutics13071010
Chicago/Turabian StyleSofia, Natalia, Stefan Mühlebach, Umberto M. Musazzi, Rani Khatib, José Manuel Martinez Sesmero, Hans-Peter Lipp, Jacqueline Surugue, Tiziana Di Francesco, and Beat Flühmann. 2021. "How Do Hospital Pharmacists Approach Substitution of Nanomedicines? Insights from a Qualitative Pilot Study and a Quantitative Market Research Analysis in Five European Countries" Pharmaceutics 13, no. 7: 1010. https://doi.org/10.3390/pharmaceutics13071010
APA StyleSofia, N., Mühlebach, S., Musazzi, U. M., Khatib, R., Martinez Sesmero, J. M., Lipp, H. -P., Surugue, J., Di Francesco, T., & Flühmann, B. (2021). How Do Hospital Pharmacists Approach Substitution of Nanomedicines? Insights from a Qualitative Pilot Study and a Quantitative Market Research Analysis in Five European Countries. Pharmaceutics, 13(7), 1010. https://doi.org/10.3390/pharmaceutics13071010