
3D-Printed Mesoporous Carrier System for Delivery of Poorly Soluble Drugs


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Mesoporous Magnesium Carbonate (MMC)
2.3. Synthesis of MCM-41
2.4. Drug Loading of Mesoporous Materials
2.5. Gas Sorption Analysis
2.6. Filament Preparation
2.7. Tablet Printing
2.8. Attenuated Total Reflectance–Fourier-Transform Infrared Spectroscopy (ATR–FTIR)
2.9. Powder X-ray Diffraction (XRD)
2.10. Differential Scanning Calorimetry (DSC)
2.11. Thermogravimetric Analysis (TGA)
2.12. Scanning Electron Microscopy (SEM)
2.13. Drug Solubility Measurement
2.14. In Vitro Release Studies
3. Results and Discussion
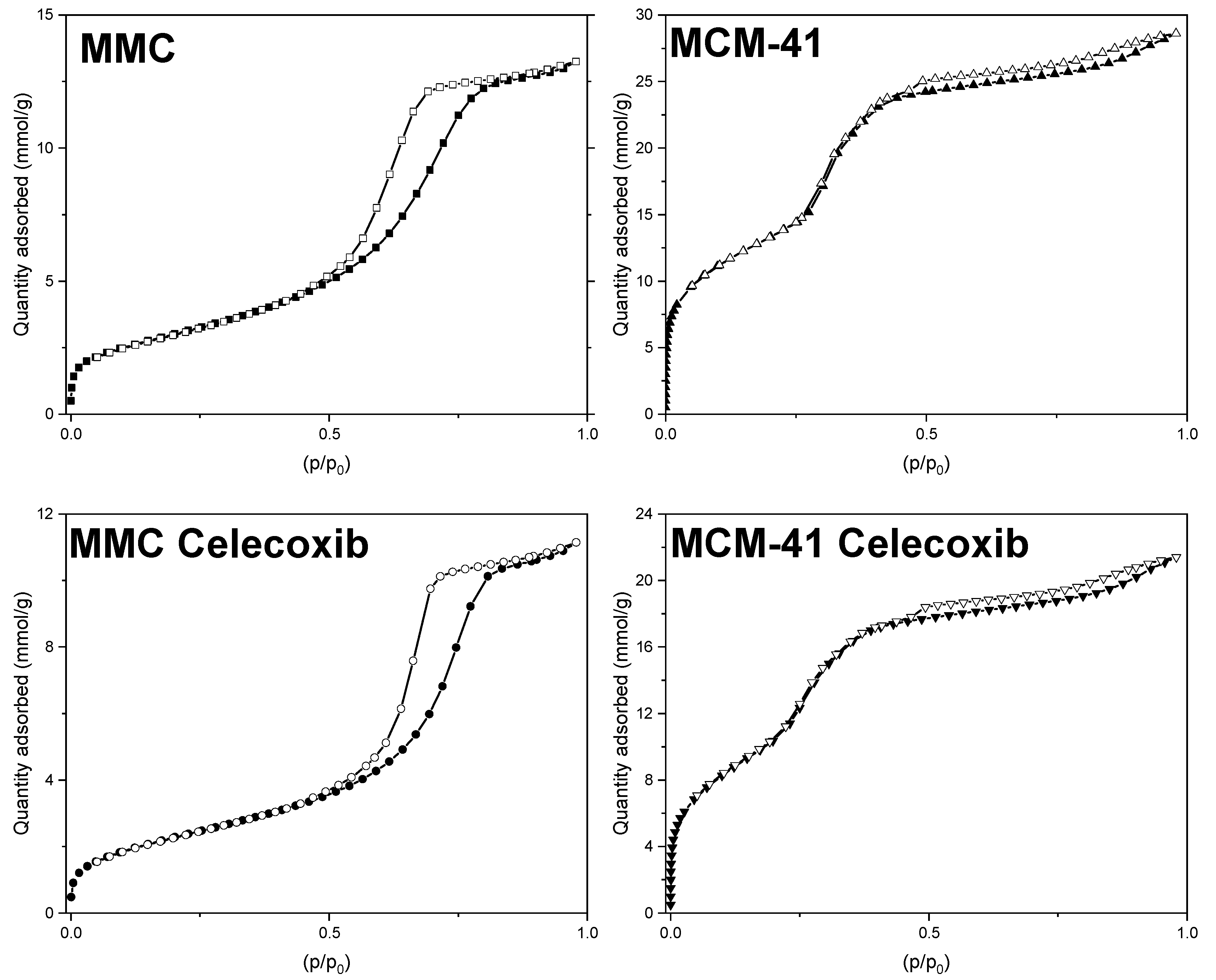
3.1. Gas Sorption Measurements
3.2. Filament Extrusion and Tablet Printing
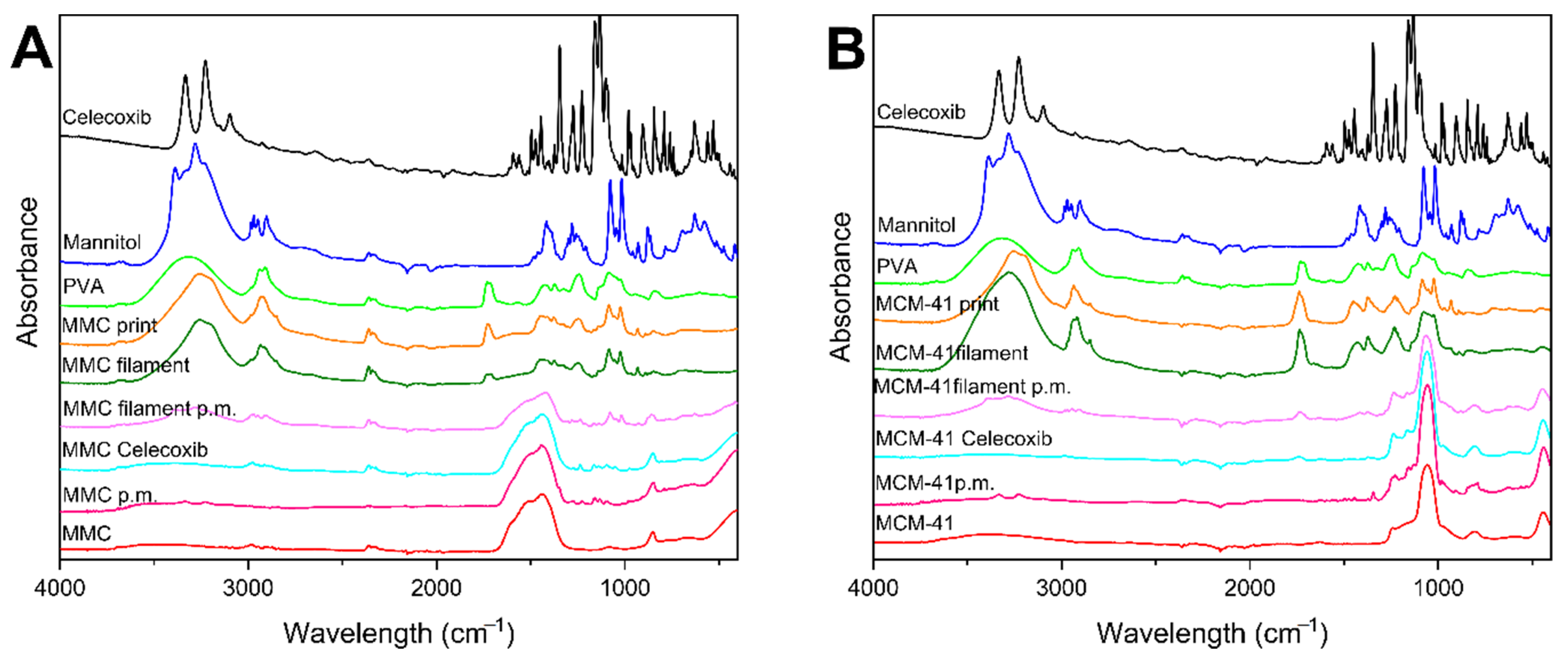
3.3. ATR–FTIR
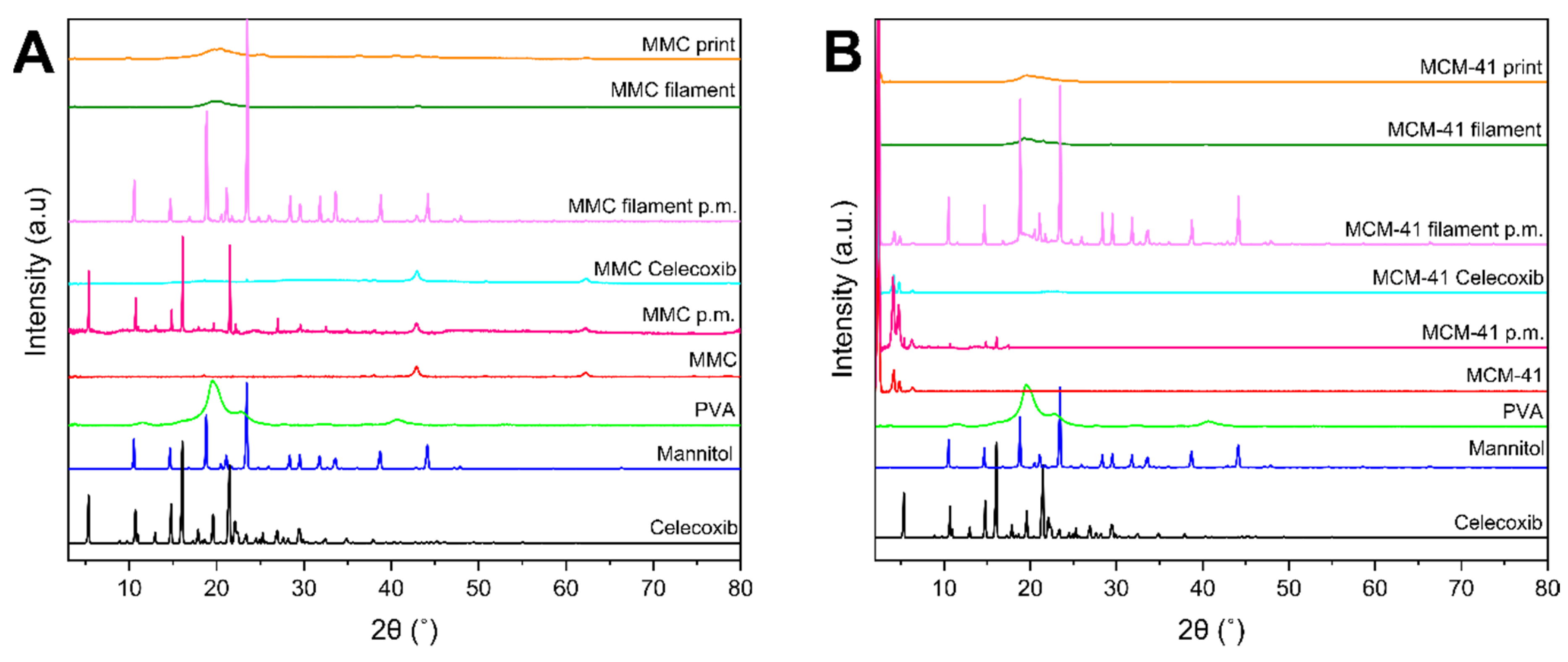
3.4. XRD
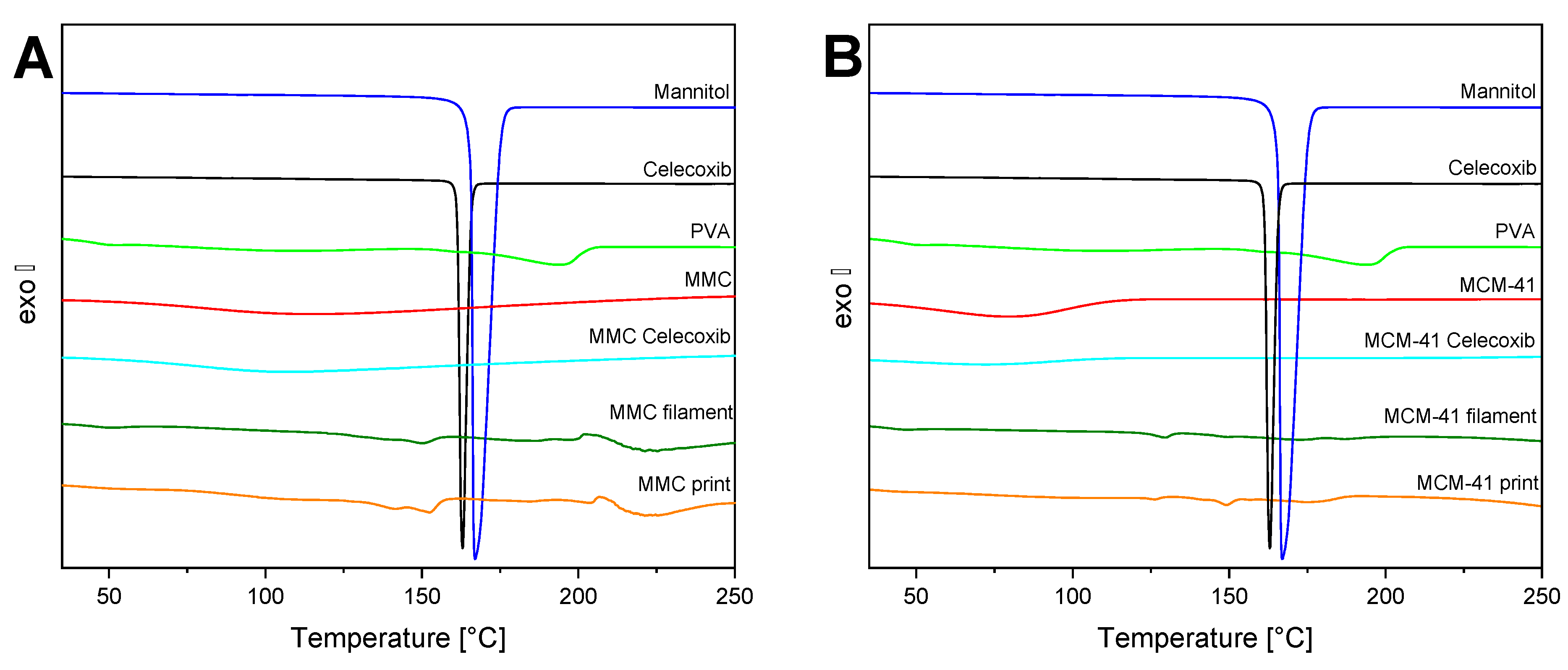
3.5. DSC
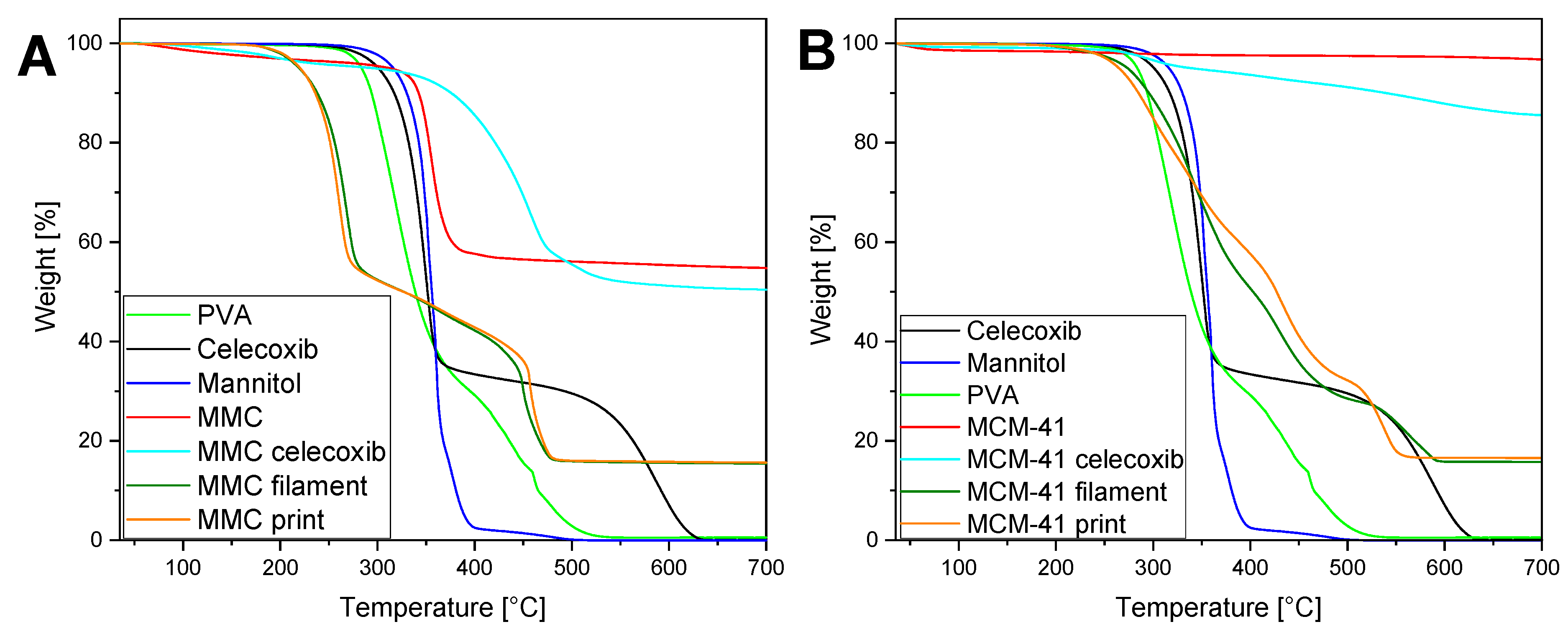
3.6. TGA
3.7. SEM
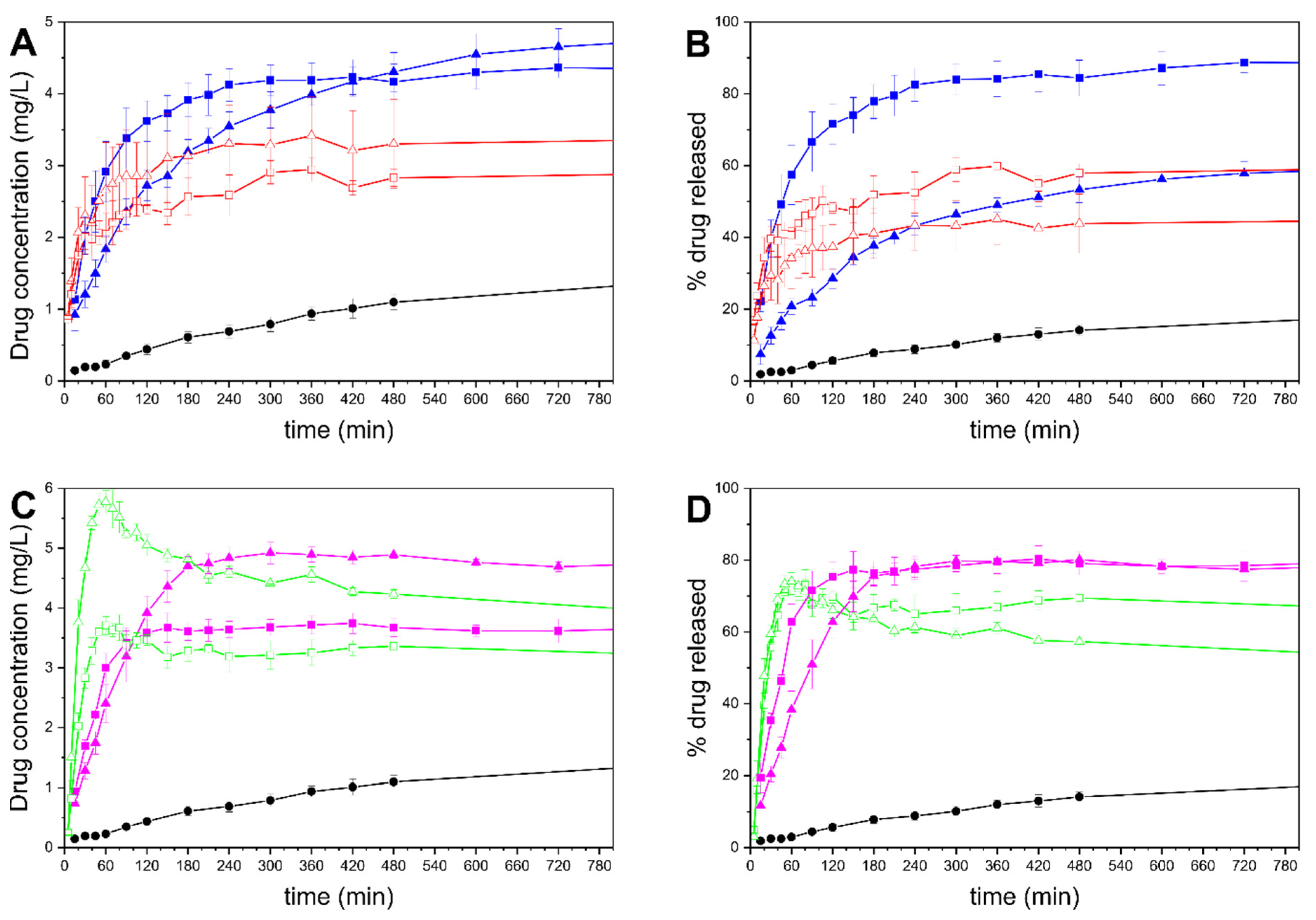
3.8. In Vitro Drug Release
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ibrahim, A.H.; Smått, J.-H.; Govardhanam, N.P.; Ibrahim, H.M.; Ismael, H.R.; Afouna, M.I.; Samy, A.M.; Rosenholm, J.M. Formulation and Optimization of Drug-Loaded Mesoporous Silica Nanoparticle-Based Tablets to Improve the Dissolution Rate of the Poorly Water-Soluble Drug Silymarin. Eur. J. Pharm. Sci. 2020, 142, 105103. [Google Scholar] [CrossRef]
- Mehta, M. Biopharmaceutics Classification System (BCS); John Wiley & Sons: New York, NY, USA, 2017; ISBN 978-1-118-47661-1. [Google Scholar]
- Jia, L. Nanoparticle Formulation Increases Oral Bioavailability of Poorly Soluble Drugs: Approaches, Experimental Evidences and Theory. Curr. Nanosci. 2005, 1, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating Drug Delivery Systems: The Answer to Solubility-Limited Oral Bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serajuddin, A.T.M. Solid Dispersion of Poorly Water-soluble Drugs: Early Promises, Subsequent Problems, and Recent Breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Bremmell, K.E.; Prestidge, C.A. Enhancing Oral Bioavailability of Poorly Soluble Drugs with Mesoporous Silica Based Systems: Opportunities and Challenges. Drug Dev. Ind. Pharm. 2019, 45, 349–358. [Google Scholar] [CrossRef]
- McCarthy, C.A.; Ahern, R.J.; Dontireddy, R.; Ryan, K.B.; Crean, A.M. Mesoporous Silica Formulation Strategies for Drug Dissolution Enhancement: A Review. Expert Opin. Drug Deliv. 2016, 13, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Van Speybroeck, M.; Mellaerts, R.; Mols, R.; Thi, T.D.; Martens, J.A.; Van Humbeeck, J.; Annaert, P.; Van den Mooter, G.; Augustijns, P. Enhanced Absorption of the Poorly Soluble Drug Fenofibrate by Tuning Its Release Rate from Ordered Mesoporous Silica. Eur. J. Pharm. Sci. 2010, 41, 623–630. [Google Scholar] [CrossRef]
- Andersson, J.; Rosenholm, J.; Areva, S.; Lindén, M. Influences of Material Characteristics on Ibuprofen Drug Loading and Release Profiles from Ordered Micro- and Mesoporous Silica Matrices. Chem. Mater. 2004, 16, 4160–4167. [Google Scholar] [CrossRef]
- Kresge, C.T.; Roth, W.J. The Discovery of Mesoporous Molecular Sieves from the Twenty Year Perspective. Chem. Soc. Rev. 2013, 42, 3663. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered Mesoporous Molecular Sieves Synthesized by a Liquid-Crystal Template Mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Bukara, K.; Schueller, L.; Rosier, J.; Daems, T.; Verheyden, L.; Eelen, S.; Martens, J.A.; Van den Mooter, G.; Bugarski, B.; Kiekens, F. In Vivo Performance of Fenofibrate Formulated With Ordered Mesoporous Silica Versus 2-Marketed Formulations: A Comparative Bioavailability Study in Beagle Dogs. J. Pharm. Sci. 2016, 105, 2381–2385. [Google Scholar] [CrossRef]
- Limnell, T.; Heikkilä, T.; Santos, H.A.; Sistonen, S.; Hellstén, S.; Laaksonen, T.; Peltonen, L.; Kumar, N.; Murzin, D.Y.; Louhi-Kultanen, M.; et al. Physicochemical Stability of High Indomethacin Payload Ordered Mesoporous Silica MCM-41 and SBA-15 Microparticles. Int. J. Pharm. 2011, 416, 242–251. [Google Scholar] [CrossRef]
- Koneru, B.; Shi, Y.; Wang, Y.-C.; Chavala, S.; Miller, M.; Holbert, B.; Conson, M.; Ni, A.; Di Pasqua, A. Tetracycline-Containing MCM-41 Mesoporous Silica Nanoparticles for the Treatment of Escherichia Coli. Molecules 2015, 20, 19690–19698. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, L.; Jiang, T.; Zhang, J.; Zhang, C.; Sun, C.; Deng, Y.; Sun, J.; Wang, S. The Investigation of MCM-48-Type and MCM-41-Type Mesoporous Silica as Oral Solid Dispersion Carriers for Water Insoluble Cilostazol. Drug Dev. Ind. Pharm. 2014, 40, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Rajput, S.J. Investigation of in Vitro Permeability and in Vivo Pharmacokinetic Behavior of Bare and Functionalized MCM-41 and MCM-48 Mesoporous Silica Nanoparticles: A Burst and Controlled Drug Release System for Raloxifene. Drug Dev. Ind. Pharm. 2019, 45, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, B.; Rámila, A.; Pérez-Pariente, J.; Díaz, I.; Vallet-Regí, M. MCM-41 Organic Modification as Drug Delivery Rate Regulator. Chem. Mater. 2003, 15, 500–503. [Google Scholar] [CrossRef]
- Datt, A.; El-Maazawi, I.; Larsen, S.C. Aspirin Loading and Release from MCM-41 Functionalized with Aminopropyl Groups via Co-Condensation or Postsynthesis Modification Methods. J. Phys. Chem. C 2012, 116, 18358–18366. [Google Scholar] [CrossRef]
- Khodaverdi, E.; Ahmadi, M.; Kamali, H.; Hadizadeh, F. Aminopropyl Groups of the Functionalized Mobil Crystalline Material 41 as a Carrier for Controlled Diclofenac Sodium and Piroxicam Delivery. Int. J. Pharm. Investig. 2017, 7, 174. [Google Scholar] [CrossRef]
- Manzano, M.; Aina, V.; Areán, C.O.; Balas, F.; Cauda, V.; Colilla, M.; Delgado, M.R.; Vallet-Regí, M. Studies on MCM-41 Mesoporous Silica for Drug Delivery: Effect of Particle Morphology and Amine Functionalization. Chem. Eng. J. 2008, 137, 30–37. [Google Scholar] [CrossRef]
- Frykstrand, S.; Forsgren, J.; Mihranyan, A.; Strømme, M. On the Pore Forming Mechanism of Upsalite, a Micro- and Mesoporous Magnesium Carbonate. Microporous Mesoporous Mater. 2014, 190, 99–104. [Google Scholar] [CrossRef]
- Forsgren, J.; Frykstrand, S.; Grandfield, K.; Mihranyan, A.; Strømme, M. A Template-Free, Ultra-Adsorbing, High Surface Area Carbonate Nanostructure. PLoS ONE 2013, 8, e68486. [Google Scholar] [CrossRef] [Green Version]
- Cheung, O.; Zhang, P.; Frykstrand, S.; Zheng, H.; Yang, T.; Sommariva, M.; Zou, X.; Strømme, M. Nanostructure and Pore Size Control of Template-Free Synthesised Mesoporous Magnesium Carbonate. RSC Adv. 2016, 6, 74241–74249. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Zardán Gómez de la Torre, T.; Forsgren, J.; Bergström, C.A.S.; Strømme, M. Diffusion-Controlled Drug Release From the Mesoporous Magnesium Carbonate Upsalite®. J. Pharm. Sci. 2016, 105, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Forsgren, J.; Strømme, M. Stabilisation of Amorphous Ibuprofen in Upsalite, a Mesoporous Magnesium Carbonate, as an Approach to Increasing the Aqueous Solubility of Poorly Soluble Drugs. Int. J. Pharm. 2014, 472, 185–191. [Google Scholar] [CrossRef]
- Yang, J.; Alvebratt, C.; Zhang, P.; Zardán Gómez de la Torre, T.; Strømme, M.; Bergström, C.A.S.; Welch, K. Enhanced Release of Poorly Water-Soluble Drugs from Synergy between Mesoporous Magnesium Carbonate and Polymers. Int. J. Pharm. 2017, 525, 183–190. [Google Scholar] [CrossRef]
- Zhang, P.; Zardán Gómez de la Torre, T.; Welch, K.; Bergström, C.; Strømme, M. Supersaturation of Poorly Soluble Drugs Induced by Mesoporous Magnesium Carbonate. Eur. J. Pharm. Sci. 2016, 93, 468–474. [Google Scholar] [CrossRef]
- Alvebratt, C.; Dening, T.J.; Åhlén, M.; Cheung, O.; Strømme, M.; Gogoll, A.; Prestidge, C.A.; Bergström, C.A.S. In Vitro Performance and Chemical Stability of Lipid-Based Formulations Encapsulated in a Mesoporous Magnesium Carbonate Carrier. Pharmaceutics 2020, 12, 426. [Google Scholar] [CrossRef] [PubMed]
- Welch, K.; Latifzada, M.A.; Frykstrand, S.; Strømme, M. Investigation of the Antibacterial Effect of Mesoporous Magnesium Carbonate. ACS Omega 2016, 1, 907–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vall, M.; Ferraz, N.; Cheung, O.; Strømme, M.; Zardán Gómez de la Torre, T. Exploring the Use of Amine Modified Mesoporous Magnesium Carbonate for the Delivery of Salicylic Acid in Topical Formulations: In Vitro Cytotoxicity and Drug Release Studies. Molecules 2019, 24, 1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, E.; Pitzanti, G.; Larrañeta, E.; Lamprou, D.A. 3D Printing of Pharmaceuticals and Drug Delivery Devices. Pharmaceutics 2020, 12, 266. [Google Scholar] [CrossRef] [Green Version]
- Gioumouxouzis, C.I.; Karavasili, C.; Fatouros, D.G. Recent Advances in Pharmaceutical Dosage Forms and Devices Using Additive Manufacturing Technologies. Drug Discov. Today 2019, 24, 636–643. [Google Scholar] [CrossRef]
- Goyanes, A.; Wang, J.; Buanz, A.; Martínez-Pacheco, R.; Telford, R.; Gaisford, S.; Basit, A.W. 3D Printing of Medicines: Engineering Novel Oral Devices with Unique Design and Drug Release Characteristics. Mol. Pharm. 2015, 12, 4077–4084. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.C.; Isreb, A.; Forbes, R.T.; Dores, F.; Habashy, R.; Petit, J.-B.; Alhnan, M.A.; Oga, E.F. ‘Temporary Plasticiser’: A Novel Solution to Fabricate 3D Printed Patient-Centred Cardiovascular ‘Polypill’ Architectures. Eur. J. Pharm. Biopharm. 2019, 135, 94–103. [Google Scholar] [CrossRef]
- Eleftheriadis, G.K.; Katsiotis, C.S.; Bouropoulos, N.; Koutsopoulos, S.; Fatouros, D.G. FDM-Printed PH-Responsive Capsules for the Oral Delivery of a Model Macromolecular Dye. Pharm. Dev. Technol. 2020, 25, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Gioumouxouzis, C.I.; Baklavaridis, A.; Katsamenis, O.L.; Markopoulou, C.K.; Bouropoulos, N.; Tzetzis, D.; Fatouros, D.G. A 3D Printed Bilayer Oral Solid Dosage Form Combining Metformin for Prolonged and Glimepiride for Immediate Drug Delivery. Eur. J. Pharm. Sci. 2018, 120, 40–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftheriadis, G.K.; Katsiotis, C.S.; Andreadis, D.A.; Tzetzis, D.; Ritzoulis, C.; Bouropoulos, N.; Kanellopoulou, D.; Andriotis, E.G.; Tsibouklis, J.; Fatouros, D.G. Inkjet Printing of a Thermolabile Model Drug onto FDM-Printed Substrates: Formulation and Evaluation. Drug Dev. Ind. Pharm. 2020, 46, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yu, X.; Jin, Y. 3D Printing of Vaginal Rings with Personalized Shapes for Controlled Release of Progesterone. Int. J. Pharm. 2018, 539, 75–82. [Google Scholar] [CrossRef]
- Persaud, S.; Eid, S.; Swiderski, N.; Serris, I.; Cho, H. Preparations of Rectal Suppositories Containing Artesunate. Pharmaceutics 2020, 12, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanki, N.G.; Tahsin, M.; Shah, A.V.; Serajuddin, A.T.M. Formulation of 3D Printed Tablet for Rapid Drug Release by Fused Deposition Modeling: Screening Polymers for Drug Release, Drug-Polymer Miscibility and Printability. J. Pharm. Sci. 2018, 107, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Gumaste, S.G.; Gupta, S.S.; Serajuddin, A.T.M. Investigation of Polymer-Surfactant and Polymer-Drug-Surfactant Miscibility for Solid Dispersion. AAPS J. 2016, 18, 1131–1143. [Google Scholar] [CrossRef]
- Govender, R.; Abrahmsén-Alami, S.; Folestad, S.; Larsson, A. High Content Solid Dispersions for Dose Window Extension: A Basis for Design Flexibility in Fused Deposition Modelling. Pharm. Res. 2020, 37, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macedo, J.; Samaro, A.; Vanhoorne, V.; Vervaet, C.; Pinto, J.F. Processability of Poly(Vinyl Alcohol) Based Filaments with Paracetamol Prepared by Hot-Melt Extrusion for Additive Manufacturing. J. Pharm. Sci. 2020, 109, 3636–3644. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, G.K.; Katsiotis, C.S.; Genina, N.; Boetker, J.; Rantanen, J.; Fatouros, D.G. Manufacturing of Hybrid Drug Delivery Systems by Utilizing the Fused Filament Fabrication (FFF) Technology. Expert Opin. Drug Deliv. 2020, 17, 1063–1068. [Google Scholar] [CrossRef]
- Eleftheriadis, G.K.; Ritzoulis, C.; Bouropoulos, N.; Tzetzis, D.; Andreadis, D.A.; Boetker, J.; Rantanen, J.; Fatouros, D.G. Unidirectional Drug Release from 3D Printed Mucoadhesive Buccal Films Using FDM Technology: In Vitro and Ex Vivo Evaluation. Eur. J. Pharm. Biopharm. 2019, 144, 180–192. [Google Scholar] [CrossRef]
- Grün, M.; Unger, K.K.; Matsumoto, A.; Tsutsumi, K. Novel Pathways for the Preparation of Mesoporous MCM-41 Materials: Control of Porosity and Morphology. Microporous Mesoporous Mater. 1999, 27, 207–216. [Google Scholar] [CrossRef]
- Abu-Diak, O.A.; Jones, D.S.; Andrews, G.P. An Investigation into the Dissolution Properties of Celecoxib Melt Extrudates: Understanding the Role of Polymer Type and Concentration in Stabilizing Supersaturated Drug Concentrations. Mol. Pharm. 2011, 8, 1362–1371. [Google Scholar] [CrossRef]
- Brunauer, S.; Deming, L.S.; Deming, W.E.; Teller, E. On a Theory of the van Der Waals Adsorption of Gases. J. Am. Chem. Soc. 1940, 62, 1723–1732. [Google Scholar] [CrossRef]
- Tzankova, V.; Aluani, D.; Yordanov, Y.; Valoti, M.; Frosini, M.; Spassova, I.; Kovacheva, D.; Tzankov, B. In Vitro Toxicity Evaluation of Lomefloxacin-Loaded MCM-41 Mesoporous Silica Nanoparticles. Drug Chem. Toxicol. 2019, 1–12. [Google Scholar] [CrossRef]
- Celecoxib|C17H14F3N3O2S|ChemSpider. Available online: http://www.chemspider.com/Chemical-Structure.2562.html (accessed on 11 January 2021).
- Wang, K. Die Swell of Complex Polymeric Systems. In Viscoelasticity—From Theory to Biological Applications; De Vicente, J., Ed.; InTech: Rijeka, Croatia, 2012; ISBN 978-953-51-0841-2. [Google Scholar]
- Gulshan, M.; Sai, M.L.S.; Hemalatha, T.; Sri, U.J.; Ramarao, N. Formulation and development of microspheres for the treatment of familial adenomatous polyposis. Int. J. Appl. Pharm. 2017, 9, 66. [Google Scholar] [CrossRef] [Green Version]
- Pandya, V.M.; Patel, D.J.; Patel, J.K.; Patel, R.P. Formulation, Characterization, and Optimization of Fast-Dissolve Tablets Containing Celecoxib Solid Dispersion. Dissolution Technol. 2009, 16, 22–27. [Google Scholar] [CrossRef]
- Kharazmi, A.; Faraji, N.; Mat Hussin, R.; Saion, E.; Yunus, W.M.M.; Behzad, K. Structural, Optical, Opto-Thermal and Thermal Properties of ZnS–PVA Nanofluids Synthesized through a Radiolytic Approach. Beilstein J. Nanotechnol. 2015, 6, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, N.V.; Nate, M.M.; Kurup, M.B.; Bambole, V.A.; Sabharwal, S. Effect of γ-Radiation on the Structure and Morphology of Polyvinyl Alcohol Films. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2005, 237, 585–592. [Google Scholar] [CrossRef]
- Al-khattawi, A.; Alyami, H.; Townsend, B.; Ma, X.; Mohammed, A.R. Evidence-Based Nanoscopic and Molecular Framework for Excipient Functionality in Compressed Orally Disintegrating Tablets. PLoS ONE 2014, 9, e101369. [Google Scholar] [CrossRef] [PubMed]
- Burger, A.; Henck, J.-O.; Hetz, S.; Rollinger, J.M.; Weissnicht, A.A.; Stöttner, H. Energy/Temperature Diagram and Compression Behavior of the Polymorphs of d-Mannitol. J. Pharm. Sci. 2000, 89, 457–468. [Google Scholar] [CrossRef]
- Guo, K.; Han, F.; Arslan, Z.; McComb, J.; Mao, X.; Zhang, R.; Sudarson, S.; Yu, H. Adsorption of Cs from Water on Surface-Modified MCM-41 Mesosilicate. Water. Air. Soil Pollut. 2015, 226, 288. [Google Scholar] [CrossRef]
- Zhu, L.; Zhou, L.; Huang, N.; Cui, W.; Liu, Z.; Xiao, K.; Zhou, Z. Efficient Preparation of Enantiopure D-Phenylalanine through Asymmetric Resolution Using Immobilized Phenylalanine Ammonia-Lyase from Rhodotorula Glutinis JN-1 in a Recirculating Packed-Bed Reactor. PLoS ONE 2014, 9, e108586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Sato, Y.; Inoue, Y. Synthesis of P(NIPAM-Co-Am)/Mesoporous Silica Composites and Their Temperature- Responsive Anion Exchange. J. Mater. Sci. Chem. Eng. 2015, 3, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Pshezhetskii, V.S.; Rakhnyanskaya, A.A.; Gaponenko, I.M.; Nalbandyan, Y.E. A Differential Scanning Calorimetry Study of Polyvinyl Alcohol. Polym. Sci. USSR 1990, 32, 722–726. [Google Scholar] [CrossRef]
- Chawla, G.; Gupta, P.; Thilagavathi, R.; Chakraborti, A.K.; Bansal, A.K. Characterization of Solid-State Forms of Celecoxib. Eur. J. Pharm. Sci. 2003, 20, 305–317. [Google Scholar] [CrossRef]
- Goyanes, A.; Robles Martinez, P.; Buanz, A.; Basit, A.W.; Gaisford, S. Effect of Geometry on Drug Release from 3D Printed Tablets. Int. J. Pharm. 2015, 494, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Kumaresan, G.; Velraj, R.; Iniyan, S. Thermal Analysis of D-Mannitol for Use as Phase Change Material for Latent Heat Storage. J. Appl. Sci. 2011, 11, 3044–3048. [Google Scholar] [CrossRef] [Green Version]
- Aydın, A.A.; Ilberg, V. Effect of Different Polyol-Based Plasticizers on Thermal Properties of Polyvinyl Alcohol:Starch Blends. Carbohydr. Polym. 2016, 136, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Barnes, T.J.; Prestidge, C.A. Celecoxib Confinement within Mesoporous Silicon for Enhanced Oral Bioavailability. Open Mater. Sci. 2014, 1. [Google Scholar] [CrossRef]
- Blaabjerg, L.; Grohganz, H.; Lindenberg, E.; Löbmann, K.; Müllertz, A.; Rades, T. The Influence of Polymers on the Supersaturation Potential of Poor and Good Glass Formers. Pharmaceutics 2018, 10, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szegedi, A.; Popova, M.; Goshev, I.; Mihály, J. Effect of Amine Functionalization of Spherical MCM-41 and SBA-15 on Controlled Drug Release. J. Solid State Chem. 2011, 184, 1201–1207. [Google Scholar] [CrossRef]
- Reynolds, T.D.; Mitchell, S.A.; Balwinski, K.M. Investigation of the Effect of Tablet Surface Area/Volume on Drug Release from Hydroxypropylmethylcellulose Controlled-Release Matrix Tablets. Drug Dev. Ind. Pharm. 2002, 28, 457–466. [Google Scholar] [CrossRef]
- Goyanes, A.; Buanz, A.B.M.; Basit, A.W.; Gaisford, S. Fused-Filament 3D Printing (3DP) for Fabrication of Tablets. Int. J. Pharm. 2014, 476, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Goyanes, A.; Buanz, A.B.M.; Hatton, G.B.; Gaisford, S.; Basit, A.W. 3D Printing of Modified-Release Aminosalicylate (4-ASA and 5-ASA) Tablets. Eur. J. Pharm. Biopharm. 2015, 89, 157–162. [Google Scholar] [CrossRef] [PubMed]









 , MMC-T50: ■, MMC-T70: ▲, MCM-41-50: □, MCM-41-70:
, MMC-T50: ■, MMC-T70: ▲, MCM-41-50: □, MCM-41-70:  , MCM-41-T50: ■, MCM-41-T70: ▲.
, MMC-T50: ■, MMC-T70: ▲, MCM-41-50: □, MCM-41-70: , MCM-41-T50: ■, MCM-41-T70: ▲.
, MCM-41-T50: ■, MCM-41-T70: ▲.
, MMC-T50: ■, MMC-T70: ▲, MCM-41-50: □, MCM-41-70: , MCM-41-T50: ■, MCM-41-T70: ▲.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystalline Celecoxib | MMC-50 | MMC-70 | MCM-41-50 | MCM-41-70 | MMC-T50 | MMC-T70 | MCM-41-T50 | MCM-41-T70 | |
|---|---|---|---|---|---|---|---|---|---|
| Celecoxib dose (mg) | 7.1 | 4.6 | 7.1 | 4.33 | 5.67 | 4.6 | 7.1 | 4.33 | 5.67 |
| Specific Surface Area (m2/g) | Pore Volume (cm3/g) | Pore Width (nm) | |
|---|---|---|---|
| MMC | 242.9 | 0.44 | 6.84 |
| MMC Celecoxib | 187.5 | 0.37 | 7.40 |
| MCM-41 | 1082.0 | 0.90 | 3.18 |
| MCM-41 Celecoxib | 840.9 | 0.68 | 2.95 |
| Mass ± St.D. (mg) | Height ± St.D. (mm) | Diameter ± St.D. (mm) | |
|---|---|---|---|
| MMC Filament | - | - | 1.60 ± 0.03 |
| MMC-T50 | 173 ± 8 | 2.96 ± 0.06 | 11.03 ± 0.03 |
| MMC-T70 | 264 ± 12 | 3.00 ± 0.01 | 11.11 ± 0.01 |
| MCM-41 Filament | - | - | 1.72 ± 0.02 |
| MCM-41-T50 | 241 ± 7 | 2.99 ± 0.02 | 11.00 ± 0.01 |
| MCM-41-T70 | 312 ± 8 | 3.00 ± 0.01 | 11.00 ± 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsiotis, C.S.; Åhlén, M.; Strømme, M.; Welch, K. 3D-Printed Mesoporous Carrier System for Delivery of Poorly Soluble Drugs. Pharmaceutics 2021, 13, 1096. https://doi.org/10.3390/pharmaceutics13071096
Katsiotis CS, Åhlén M, Strømme M, Welch K. 3D-Printed Mesoporous Carrier System for Delivery of Poorly Soluble Drugs. Pharmaceutics. 2021; 13(7):1096. https://doi.org/10.3390/pharmaceutics13071096
Chicago/Turabian StyleKatsiotis, Christos S., Michelle Åhlén, Maria Strømme, and Ken Welch. 2021. "3D-Printed Mesoporous Carrier System for Delivery of Poorly Soluble Drugs" Pharmaceutics 13, no. 7: 1096. https://doi.org/10.3390/pharmaceutics13071096
APA StyleKatsiotis, C. S., Åhlén, M., Strømme, M., & Welch, K. (2021). 3D-Printed Mesoporous Carrier System for Delivery of Poorly Soluble Drugs. Pharmaceutics, 13(7), 1096. https://doi.org/10.3390/pharmaceutics13071096