

Prevention and Eradication of Biofilm by Dendrimers: A Possibility Still Little Explored



Abstract
:1. Introduction to Microbial Resistance
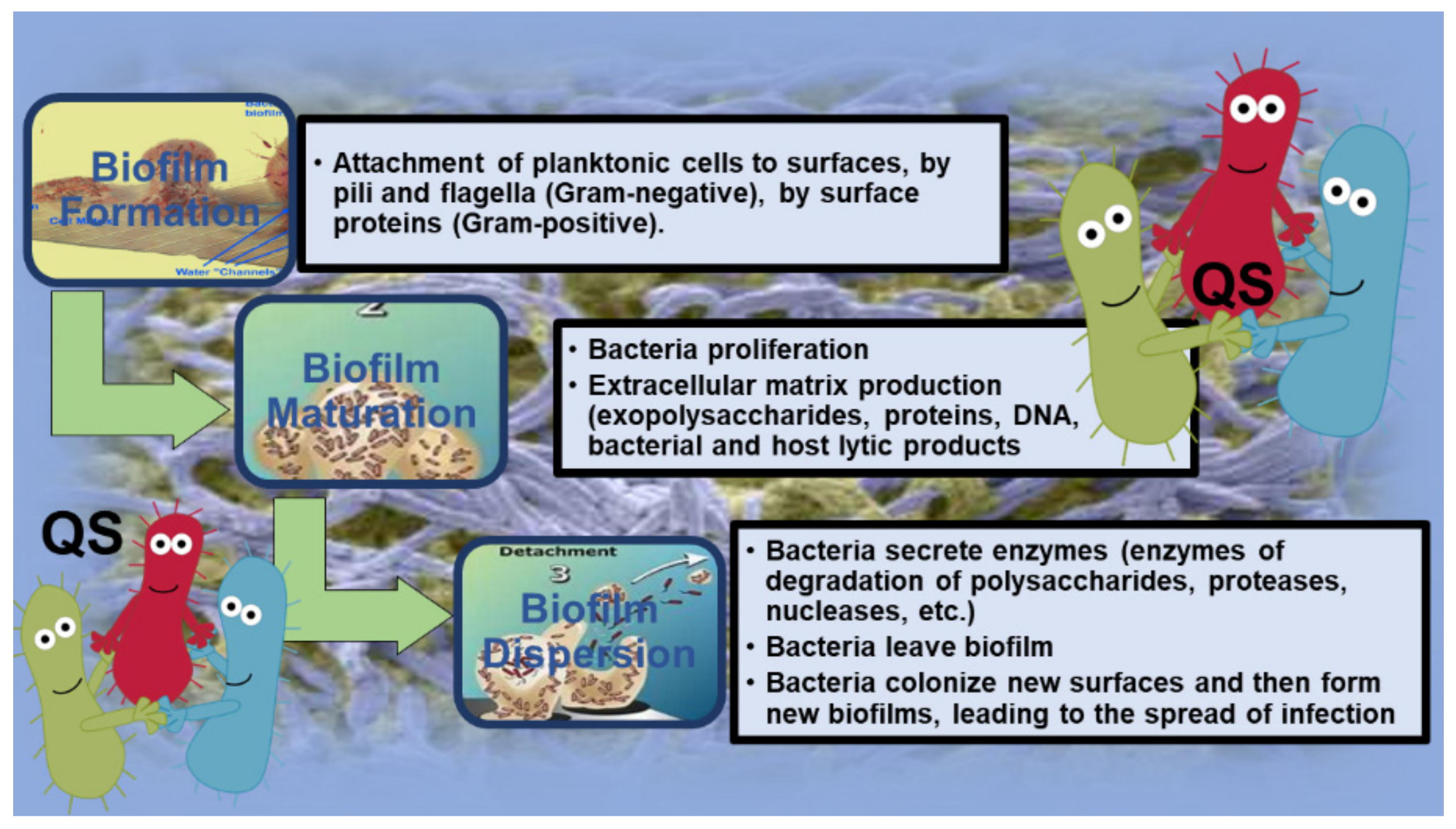
2. Introduction to Biofilm (BF)
2.1. BFs by P. aeruginosa
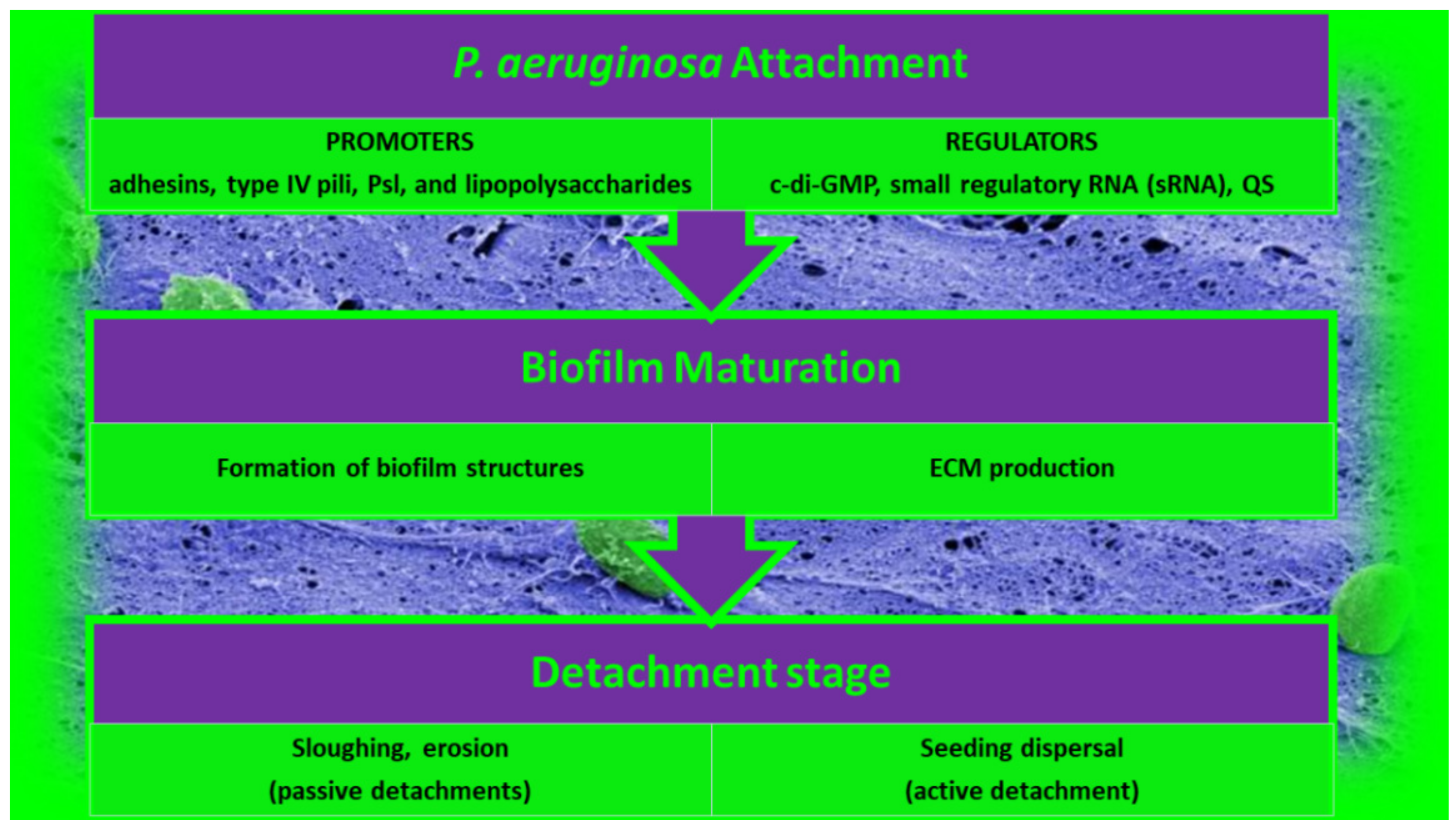
2.1.1. Attachment of P. aeruginosa BFs
2.1.2. Maturation of P. aeruginosa BFs
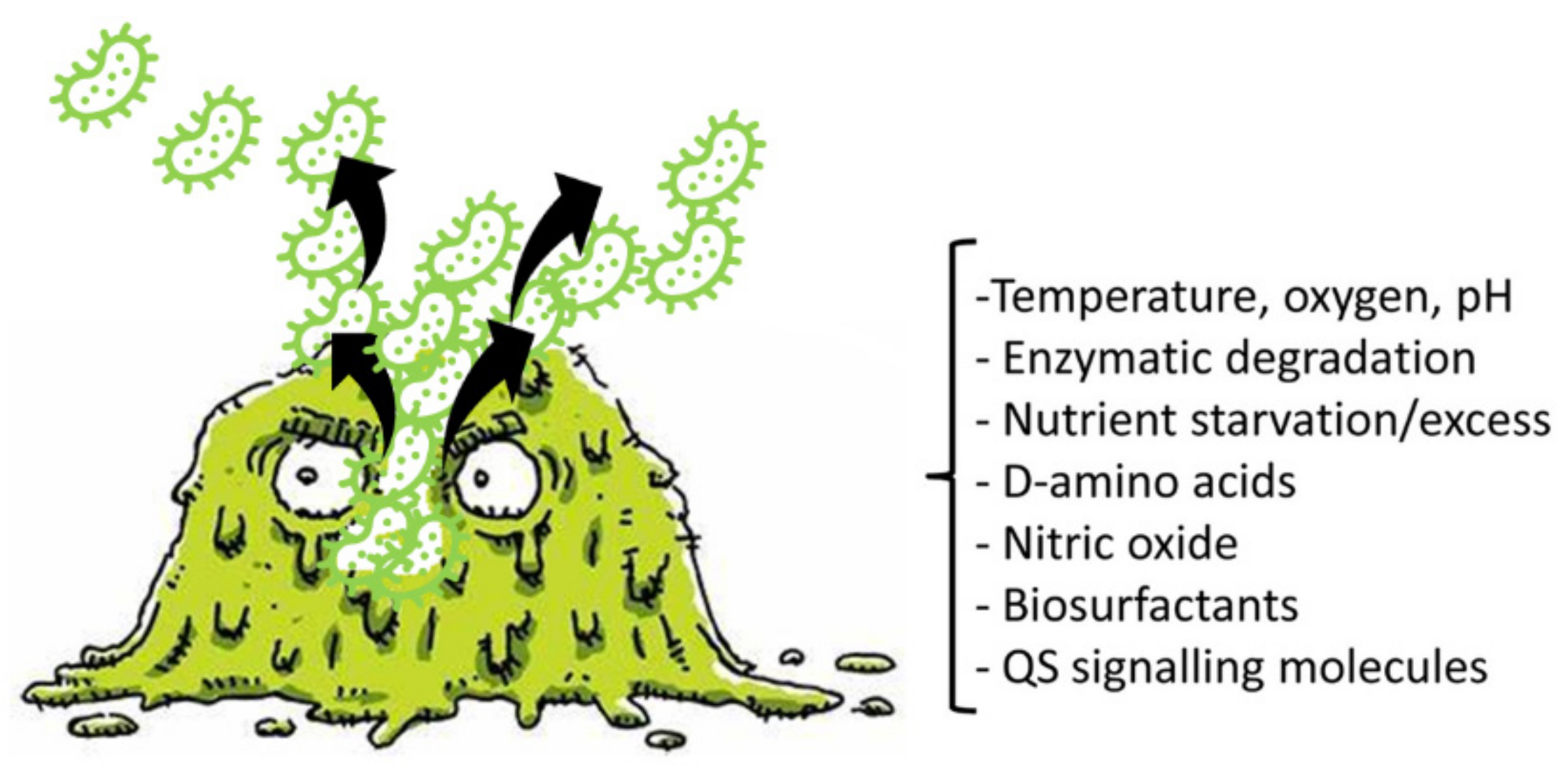
2.1.3. Detachment of P. aeruginosa BFs
2.1.4. Important Characteristics of P. aeruginosa BF
2.1.5. P. aeruginosa BF-Associated Infections
3. Prevention and/or Eradication of BF by Dendrimers: A Possibility Still Little Explored
3.1. Antibacterial Cationic Macromolecules
Cationic Dendrimers
3.2. Recently Reported Case Studies
3.3. Promising Areas for Modifying Dendrimer Matrices
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vadivelu, J. Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education; Formatex Research Center: Copenhagen, Denmark, 2013. [Google Scholar]
- Popęda, M.; Płuciennik, E.; Bednarek, A.K. Proteins in cancer resistance. Postępy Hig. Med. Do’swiadczalnej 2014, 68, 616–632. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-Resistant, Extensively Drug-Resistant and Pandrug-Resistant Bacteria: An International Expert Proposal for Interim Standard Definitions for Acquired Resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Caviglia, D.; Zorzoli, A.; Marimpietri, D.; Spallarossa, A.; Lusardi, M.; Zuccari, G.; Schito, A.M. Potent and Broad-Spectrum Bactericidal Activity of a Nanotechnologically Manipulated Novel Pyrazole. Biomedicines 2022, 10, 907. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Multidrug Resistance in Bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Antimicrobial Resistance Surveillance System (GLASS) Report: Early Implementation 2016–2017; World Health Organization: Geneva, Switzerland, 2017; ISBN 978-92-4-151344-9. [Google Scholar]
- Loeffler, J.; Stevens, D.A. Antifungal drug resistance. Clin. Infect. Dis. 2003, 36 (Suppl. 1), S31–S41. [Google Scholar] [CrossRef] [PubMed]
- Rodero, L.; Mellado, E.; Rodriguez, A.C.; Salve, A.; Guelfand, L.; Cahn, P.; Cuenca-Estrella, M.; Davel, G.; Rodriguez-Tudela, J.L. G484S Amino Acid Substitution in Lanosterol 14-α Demethylase (ERG11) Is Related to Fluconazole Resistance in a Recurrent Cryptococcus neoformans Clinical Isolate. Antimicrob. Agents Chemother. 2003, 47, 3653–3656. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.J.; Arendrup, M.C. Acquired Antifungal Drug Resistance in Aspergillus Fumigatus: Epidemiology and Detection. Med. Mycol. 2011, 49, S90–S95. [Google Scholar] [CrossRef]
- Cuenca-Estrella, M.; Gomez-Lopez, A.; Mellado, E.; Buitrago, M.J.; Monzón, A.; Rodriguez-Tudela, J.L. Scopulariopsis brevicaulis, a Fungal Pathogen Resistant to Broad-Spectrum Antifungal Agents. Antimicrob. Agents Chemother. 2003, 47, 2339–2341. [Google Scholar] [CrossRef]
- Lurain, N.S.; Chou, S. Antiviral Drug Resistance of Human Cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef]
- Wutzler, P. Antiviral Therapy of Herpes Simplex and Varicella-Zoster Virus Infections. Intervirology 1997, 40, 343–356. [Google Scholar] [CrossRef]
- Cortez, K.J.; Maldarelli, F. Clinical Management of HIV Drug Resistance. Viruses 2011, 3, 347–378. [Google Scholar] [CrossRef]
- Hurt, A.C. The Epidemiology and Spread of Drug Resistant Human Influenza Viruses. Curr. Opin. Virol. 2014, 8, 22–29. [Google Scholar] [CrossRef]
- Suppiah, J.; Mohd Zain, R.; Haji Nawi, S.; Bahari, N.; Saat, Z. Drug-Resistance Associated Mutations in Polymerase (P) Gene of Hepatitis B Virus Isolated from Malaysian HBV Carriers. Hepat. Mon. 2014, 14, e13173. [Google Scholar] [CrossRef]
- Bloland, P.B.; World Health Organization. Anti-Infective Drug Resistance Surveillance and Containment Team. Drug Resistance in Malaria. 2001. Available online: https://apps.who.int/iris/handle/10665/66847 (accessed on 19 August 2022).
- Vanaerschot, M.; Dumetz, F.; Roy, S.; Ponte-Sucre, A.; Arevalo, J.; Dujardin, J.-C. Treatment Failure in Leishmaniasis: Drug-Resistance or Another (Epi-) Phenotype? Expert Rev. Anti-Infect. Ther. 2014, 12, 937–946. [Google Scholar] [CrossRef]
- Mohapatra, S. Drug Resistance in Leishmaniasis: Newer Developments. Trop. Parasitol. 2014, 4, 4–9. [Google Scholar] [CrossRef]
- Fallon, P.G.; Doenhoff, M.J. Drug-Resistant Schistosomiasis: Resistance to Praziquantel and Oxamniquine Induced in Schistosoma mansoni in Mice Is Drug Specific. Am. J. Trop. Med. Hyg. 1994, 51, 83–88. [Google Scholar] [CrossRef]
- Qi, L.; Cui, J. A Schistosomiasis Model with Praziquantel Resistance. Discret. Dyn. Nat. Soc. 2013, 2013, e945767. [Google Scholar] [CrossRef]
- Bansal, D.; Malla, N.; Mahajan, R. Drug Resistance in Amoebiasis. Indian J. Med. Res. 2006, 123, 115–118. [Google Scholar]
- Muzny, C.A.; Schwebke, J.R. The Clinical Spectrum of Trichomonas Vaginalis Infection and Challenges to Management. Sex. Transm. Infect. 2013, 89, 423–425. [Google Scholar] [CrossRef]
- McFadden, D.C.; Tomavo, S.; Berry, E.A.; Boothroyd, J.C. Characterization of Cytochrome b from Toxoplasma gondii and Qo Domain Mutations as a Mechanism of Atovaquone-Resistance. Mol. Biochem. Parasitol. 2000, 108, 1–12. [Google Scholar] [CrossRef]
- Nagamune, K.; Moreno, S.N.J.; Sibley, L.D. Artemisinin-Resistant Mutants of Toxoplasma gondii Have Altered Calcium Homeostasis. Antimicrob. Agents Chemother. 2007, 51, 3816–3823. [Google Scholar] [CrossRef] [Green Version]
- Doliwa, C.; Escotte-Binet, S.; Aubert, D.; Sauvage, V.; Velard, F.; Schmid, A.; Villena, I. Sulfadiazine Resistance in Toxoplasma gondii: No Involvement of Overexpression or Polymorphisms in Genes of Therapeutic Targets and ABC Transporters. Parasite 2013, 20, 19. [Google Scholar] [CrossRef]
- Ullman, B. Multidrug Resistance and P-Glycoproteins in Parasitic Protozoa. J. Bioenerg. Biomembr. 1995, 27, 77–84. [Google Scholar] [CrossRef]
- Greenberg, R.M. New Approaches for Understanding Mechanisms of Drug Resistance in Schistosomes. Parasitology 2013, 140, 1534–1546. [Google Scholar] [CrossRef]
- Stoodley, P.; Sauer, K.; Davies, D.G.; Costerton, J.W. Biofilms as Complex Differentiated Communities. Annu. Rev. Microbiol. 2002, 56, 187–209. [Google Scholar] [CrossRef]
- Dincer, S.; Özdenefe, M.S.; Arkut, A. Bacterial Biofilms; BoD—Books on Demand: Norderstedt, Germany, 2020; ISBN 978-1-78985-899-0. [Google Scholar]
- Kostakioti, M.; Hadjifrangiskou, M.; Hultgren, S.J. Bacterial Biofilms: Development, Dispersal, and Therapeutic Strategies in the Dawn of the Postantibiotic Era. Cold Spring Harb. Perspect. Med. 2013, 3, a010306. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Ciofu, O.; Molin, S.; Givskov, M.; Høiby, N. Applying Insights from Biofilm Biology to Drug Development—Can a New Approach Be Developed? Nat. Rev. Drug Discov. 2013, 12, 791–808. [Google Scholar] [CrossRef]
- O’Toole, G.; Kaplan, H.B.; Kolter, R. Biofilm Formation as Microbial Development. Annu. Rev. Microbiol. 2000, 54, 49–79. [Google Scholar] [CrossRef]
- Cucarella, C.; Solano, C.; Valle, J.; Amorena, B.; Lasa, Í.; Penadés, J.R. Bap, a Staphylococcus aureus Surface Protein Involved in Biofilm Formation. J. Bacteriol. 2001, 183, 2888–2896. [Google Scholar] [CrossRef]
- Branda, S.S.; Vik, Å.; Friedman, L.; Kolter, R. Biofilms: The Matrix Revisited. Trends Microbiol. 2005, 13, 20–26. [Google Scholar] [CrossRef]
- Costerton, W.; Veeh, R.; Shirtliff, M.; Pasmore, M.; Post, C.; Ehrlich, G. The Application of Biofilm Science to the Study and Control of Chronic Bacterial Infections. J. Clin. Invest. 2003, 112, 1466–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, H.-S.; Otto, M. Molecular Basis of In Vivo Biofilm Formation by Bacterial Pathogens. Chem. Biol. 2012, 19, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, R. Biofilms: Microbial cities of scientific significance. J. Microbiol. Exp. 2014, 1, 84–98. [Google Scholar] [CrossRef]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial Biofilm and Associated Infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef]
- Hathroubi, S.; Mekni, M.A.; Domenico, P.; Nguyen, D.; Jacques, M. Biofilms: Microbial Shelters Against Antibiotics. Microb. Drug Resist. 2017, 23, 147–156. [Google Scholar] [CrossRef]
- Gebreyohannes, G.; Nyerere, A.; Bii, C.; Sbhatu, D.B. Challenges of Intervention, Treatment, and Antibiotic Resistance of Biofilm-Forming Microorganisms. Heliyon 2019, 5, e02192. [Google Scholar] [CrossRef]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial Biofilms: A Common Cause of Persistent Infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef]
- Costerton, J.W.; Cheng, K.J.; Geesey, G.G.; Ladd, T.I.; Nickel, J.C.; Dasgupta, M.; Marrie, T.J. Bacterial Biofilms in Nature and Disease. Annu. Rev. Microbiol. 1987, 41, 435–464. [Google Scholar] [CrossRef]
- Costerton, J.W.; Montanaro, L.; Arciola, C.r. Bacterial Communications in Implant Infections: A Target for an Intelligence War. Int. J. Artif. Organs 2007, 30, 757–763. [Google Scholar] [CrossRef]
- Wolcott, R.d.; Rhoads, D.d.; Bennett, M.e.; Wolcott, B.m.; Gogokhia, L.; Costerton, J.w.; Dowd, S.e. Chronic Wounds and the Medical Biofilm Paradigm. J. Wound Care 2010, 19, 45–53. [Google Scholar] [CrossRef]
- James, G.A.; Swogger, E.; Wolcott, R.; deLancey Pulcini, E.; Secor, P.; Sestrich, J.; Costerton, J.W.; Stewart, P.S. Biofilms in Chronic Wounds. Wound Repair Regen. 2008, 16, 37–44. [Google Scholar] [CrossRef]
- Bjarnsholt, T. The Role of Bacterial Biofilms in Chronic Infections. APMIS 2013, 121, 1–58. [Google Scholar] [CrossRef]
- Gjødsbøl, K.; Christensen, J.J.; Karlsmark, T.; Jørgensen, B.; Klein, B.M.; Krogfelt, K.A. Multiple Bacterial Species Reside in Chronic Wounds: A Longitudinal Study. Int. Wound J. 2006, 3, 225–231. [Google Scholar] [CrossRef]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef]
- American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am. J. Respir. Crit. Care Med. 2005, 171, 388–416. [Google Scholar] [CrossRef]
- Oliver, A.; Cantón, R.; Campo, P.; Baquero, F.; Blázquez, J. High Frequency of Hypermutable Pseudomonas aeruginosa in Cystic Fibrosis Lung Infection. Science 2000, 288, 1251–1253. [Google Scholar] [CrossRef]
- Maciá, M.D.; Blanquer, D.; Togores, B.; Sauleda, J.; Pérez, J.L.; Oliver, A. Hypermutation Is a Key Factor in Development of Multiple-Antimicrobial Resistance in Pseudomonas aeruginosa Strains Causing Chronic Lung Infections. Antimicrob. Agents Chemother. 2005, 49, 3382–3386. [Google Scholar] [CrossRef]
- Herrmann, M.; Vaudaux, P.E.; Pittet, D.; Auckenthaler, R.; Lew, P.D.; Perdreau, F.S.; Peters, G.; Waldvogel, F.A. Fibronectin, Fibrinogen, and Laminin Act as Mediators of Adherence of Clinical Staphylococcal Isolates to Foreign Material. J. Infect. Dis. 1988, 158, 693–701. [Google Scholar] [CrossRef]
- Nobile, C.J.; Mitchell, A.P. Genetics and Genomics of Candida albicans Biofilm Formation. Cell. Microbiol. 2006, 8, 1382–1391. [Google Scholar] [CrossRef]
- Nobile, C.J.; Johnson, A.D. Candida albicans Biofilms and Human Disease. Annu. Rev. Microbiol. 2015, 69, 71–92. [Google Scholar] [CrossRef]
- Gökmenoğlu, C.; Kara, N.B.; Beldüz, M.; Kamburoğlu, A.; Tosun, İ.; Sadik, E.; Kara, C. Evaluation of Candida albicans Biofilm Formation on Various Parts of Implant Material Surfaces. Niger. J. Clin. Pract. 2018, 21, 33–37. [Google Scholar] [CrossRef]
- Bouza, E.; Guinea, J.; Guembe, M. The Role of Antifungals against Candida Biofilm in Catheter-Related Candidemia. Antibiotics 2015, 4, 1–17. [Google Scholar] [CrossRef]
- Uppuluri, P.; Pierce, C.G.; López-Ribot, J.L. Candida albicans Biofilm Formation and Its Clinical Consequences. Future Microbiol. 2009, 4, 1235–1237. [Google Scholar] [CrossRef]
- Gulati, M.; Nobile, C.J. Candida albicans Biofilms: Development, Regulation, and Molecular Mechanisms. Microbes Infect. 2016, 18, 310–321. [Google Scholar] [CrossRef]
- Lara, H.H.; Romero-Urbina, D.G.; Pierce, C.; Lopez-Ribot, J.L.; Arellano-Jiménez, M.J.; Jose-Yacaman, M. Effect of Silver Nanoparticles on Candida albicans Biofilms: An Ultrastructural Study. J. Nanobiotechnol. 2015, 13, 91. [Google Scholar] [CrossRef]
- Heredero-Bermejo, I.; Gómez-Casanova, N.; Quintana, S.; Soliveri, J.; de la Mata, F.J.; Pérez-Serrano, J.; Sánchez-Nieves, J.; Copa-Patiño, J.L. In Vitro Activity of Carbosilane Cationic Dendritic Molecules on Prevention and Treatment of Candida albicans Biofilms. Pharmaceutics 2020, 12, 918. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, Y.; Wang, H.; Brash, J.; Chen, H. Anti-Fouling Bioactive Surfaces. Acta Biomater. 2011, 7, 1550–1557. [Google Scholar] [CrossRef]
- Wei, T.; Yu, Q.; Chen, H. Antibacterial Coatings: Responsive and Synergistic Antibacterial Coatings: Fighting against Bacteria in a Smart and Effective Way. Adv. Healthc. Mater. 2019, 8, 1970007. [Google Scholar] [CrossRef]
- Alfei, S.; Schito, A.M. From Nanobiotechnology, Positively Charged Biomimetic Dendrimers as Novel Antibacterial Agents: A Review. Nanomaterials 2020, 10, 2022. [Google Scholar] [CrossRef]
- Alfei, S.; Grazia Signorello, M.; Schito, A.; Catena, S.; Turrini, F. Reshaped as Polyester-Based Nanoparticles, Gallic Acid Inhibits Platelet Aggregation, Reactive Oxygen Species Production and Multi-Resistant Gram-Positive Bacteria with an Efficiency Never Obtained. Nanoscale Adv. 2019, 1, 4148–4157. [Google Scholar] [CrossRef]
- Alfei, S.; Marengo, B.; Domenicotti, C. Polyester-Based Dendrimer Nanoparticles Combined with Etoposide Have an Improved Cytotoxic and Pro-Oxidant Effect on Human Neuroblastoma Cells. Antioxidants 2020, 9, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfei, S.; Catena, S.; Turrini, F. Biodegradable and Biocompatible Spherical Dendrimer Nanoparticles with a Gallic Acid Shell and a Double-Acting Strong Antioxidant Activity as Potential Device to Fight Diseases from “Oxidative Stress”. Drug Deliv. Transl. Res. 2020, 10, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Marengo, B.; Zuccari, G.; Turrini, F.; Domenicotti, C. Dendrimer Nanodevices and Gallic Acid as Novel Strategies to Fight Chemoresistance in Neuroblastoma Cells. Nanomaterials 2020, 10, 1243. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Brullo, C.; Caviglia, D.; Piatti, G.; Zorzoli, A.; Marimpietri, D.; Zuccari, G.; Schito, A.M. Pyrazole-Based Water-Soluble Dendrimer Nanoparticles as a Potential New Agent against Staphylococci. Biomedicines 2022, 10, 17. [Google Scholar] [CrossRef]
- Schito, A.M.; Caviglia, D.; Piatti, G.; Zorzoli, A.; Marimpietri, D.; Zuccari, G.; Schito, G.C.; Alfei, S. Efficacy of Ursolic Acid-Enriched Water-Soluble and Not Cytotoxic Nanoparticles against Enterococci. Pharmaceutics 2021, 13, 1976. [Google Scholar] [CrossRef]
- Schito, A.M.; Schito, G.C.; Alfei, S. Synthesis and Antibacterial Activity of Cationic Amino Acid-Conjugated Dendrimers Loaded with a Mixture of Two Triterpenoid Acids. Polymers 2021, 13, 521. [Google Scholar] [CrossRef]
- Schito, A.M.; Alfei, S. Antibacterial Activity of Non-Cytotoxic, Amino Acid-Modified Polycationic Dendrimers against Pseudomonas aeruginosa and Other Non-Fermenting Gram-Negative Bacteria. Polymers 2020, 12, 1818. [Google Scholar] [CrossRef]
- Alfei, S.; Caviglia, D.; Piatti, G.; Zuccari, G.; Schito, A.M. Bactericidal Activity of a Self-Biodegradable Lysine-Containing Dendrimer against Clinical Isolates of Acinetobacter Genus. Int. J. Mol. Sci. 2021, 22, 7274. [Google Scholar] [CrossRef]
- Polcyn, P.; Jurczak, M.; Rajnisz, A.; Solecka, J.; Urbanczyk-Lipkowska, Z. Design of Antimicrobially Active Small Amphiphilic Peptide Dendrimers. Molecules 2009, 14, 3881–3905. [Google Scholar] [CrossRef]
- Janiszewska, J.; Sowińska, M.; Rajnisz, A.; Solecka, J.; Łącka, I.; Milewski, S.; Urbańczyk-Lipkowska, Z. Novel Dendrimeric Lipopeptides with Antifungal Activity. Bioorg. Med. Chem. Lett. 2012, 22, 1388–1393. [Google Scholar] [CrossRef]
- Winnicka, K.; Wroblewska, M.; Wieczorek, P.; Sacha, P.T.; Tryniszewska, E. Hydrogel of Ketoconazole and PAMAM Dendrimers: Formulation and Antifungal Activity. Molecules 2012, 17, 4612–4624. [Google Scholar] [CrossRef]
- Stolarska, M.; Gucwa, K.; Urbańczyk-Lipkowska, Z.; Andruszkiewicz, R. Peptide Dendrimers as Antifungal Agents and Carriers for Potential Antifungal Agent—N3-(4-Methoxyfumaroyl)-(S)-2,3-Diaminopropanoic Acid—Synthesis and Antimicrobial Activity. J. Pept. Sci. 2020, 26, e3226. [Google Scholar] [CrossRef]
- Gómez-Casanova, N.; Copa-Patiño, J.L.; Heredero-Bermejo, I. Novel Treatment Approach against Candida spp.: Evaluation of Antifungal and Antibiofilm In Vitro Activity of Dendritic Molecules. In Candida and Candidiasis; IntechOpen: London, UK, 2022. [Google Scholar] [CrossRef]
- Fuentes-Paniagua, E.; Sánchez-Nieves, J.; Hernández-Ros, J.M.; Fernández-Ezequiel, A.; Soliveri, J.; Copa-Patiño, J.L.; Gómez, R.; de la Mata, F.J. Structure–Activity Relationship Study of Cationic Carbosilane Dendritic Systems as Antibacterial Agents. RSC Adv. 2016, 6, 7022–7033. [Google Scholar] [CrossRef]
- Winnicka, K.; Sosnowska, K.; Wieczorek, P.; Sacha, P.T.; Tryniszewska, E. Poly(Amidoamine) Dendrimers Increase Antifungal Activity of Clotrimazole. Biol. Pharm. Bull. 2011, 34, 1129–1133. [Google Scholar] [CrossRef]
- Gorain, B.; Pandey, M.; Choudhury, H.; Jain, G.K.; Kesharwani, P. Chapter 15—Dendrimer for Solubility Enhancement. In Dendrimer-Based Nanotherapeutics; Kesharwani, P., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 273–283. ISBN 978-0-12-821250-9. [Google Scholar]
- Verstraeten, N.; Braeken, K.; Debkumari, B.; Fauvart, M.; Fransaer, J.; Vermant, J.; Michiels, J. Living on a surface: Swarming and biofilm formation. Trends Microbiol. 2008, 16, 496–506. [Google Scholar] [CrossRef]
- Tolker-Nielsen, T. Biofilm Development. Microbiol. Spectr. 2015, 3, MB-0001-2014. [Google Scholar] [CrossRef]
- Gjermansen, M.; Nilsson, M.; Yang, L.; Tolker-Nielsen, T. Characterization of Starvation-Induced Dispersion in Pseudomonas putida Biofilms: Genetic Elements and Molecular Mechanisms. Mol. Microbiol. 2010, 75, 815–826. [Google Scholar] [CrossRef]
- Tischler, A.D.; Camilli, A. Cyclic Diguanylate (c-Di-GMP) Regulates Vibrio Cholerae Biofilm Formation. Mol. Microbiol. 2004, 53, 857–869. [Google Scholar] [CrossRef]
- Lim, B.; Beyhan, S.; Meir, J.; Yildiz, F.H. Cyclic-DiGMP Signal Transduction Systems in Vibrio cholerae: Modulation of Rugosity and Biofilm Formation. Mol. Microbiol. 2006, 60, 331–348. [Google Scholar] [CrossRef]
- Hickman, J.W.; Tifrea, D.F.; Harwood, C.S. A Chemosensory System That Regulates Biofilm Formation through Modulation of Cyclic Diguanylate Levels. Proc. Natl. Acad. Sci. USA 2005, 102, 14422–14427. [Google Scholar] [CrossRef] [Green Version]
- Kulesekara, H.; Lee, V.; Brencic, A.; Liberati, N.; Urbach, J.; Miyata, S.; Lee, D.G.; Neely, A.N.; Hyodo, M.; Hayakawa, Y.; et al. Analysis of Pseudomonas aeruginosa Diguanylate Cyclases and Phosphodiesterases Reveals a Role for Bis-(3′-5′)-Cyclic-GMP in Virulence. Proc. Natl. Acad. Sci. USA 2006, 103, 2839–2844. [Google Scholar] [CrossRef]
- Petrova, O.E.; Sauer, K. A Novel Signaling Network Essential for Regulating Pseudomonas aeruginosa Biofilm Development. PLoS Pathog. 2009, 5, e1000668. [Google Scholar] [CrossRef]
- Petrova, O.E.; Sauer, K. SagS Contributes to the Motile-Sessile Switch and Acts in Concert with BfiSR to Enable Pseudomonas aeruginosa Biofilm Formation. J. Bacteriol. 2011, 193, 6614–6628. [Google Scholar] [CrossRef]
- Colvin, K.M.; Irie, Y.; Tart, C.S.; Urbano, R.; Whitney, J.C.; Ryder, C.; Howell, P.L.; Wozniak, D.J.; Parsek, M.R. The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ. Microbiol. 2012, 14, 1913–1928. [Google Scholar] [CrossRef]
- Kim, S.-K.; Lee, J.-H. Biofilm Dispersion in Pseudomonas aeruginosa. J. Microbiol. 2016, 54, 71–85. [Google Scholar] [CrossRef]
- Harmsen, M.; Yang, L.; Pamp, S.J.; Tolker-Nielsen, T. An Update on Pseudomonas aeruginosa Biofilm Formation, Tolerance, and Dispersal. FEMS Immunol. Med. Microbiol. 2010, 59, 253–268. [Google Scholar] [CrossRef]
- Fullagar, J.L.; Garner, A.L.; Struss, A.K.; Day, J.A.; Martin, D.P.; Yu, J.; Cai, X.; Janda, K.D.; Cohen, S.M. Antagonism of a Zinc Metalloprotease Using a Unique Metal-Chelating Scaffold: Tropolones as Inhibitors of P. aeruginosa Elastase. Chem. Commun. 2013, 49, 3197–3199. [Google Scholar] [CrossRef]
- Oglesby-Sherrouse, A.G.; Djapgne, L.; Nguyen, A.T.; Vasil, A.I.; Vasil, M.L. The Complex Interplay of Iron, Biofilm Formation, and Mucoidy Affecting Antimicrobial Resistance of Pseudomonas aeruginosa. Pathog. Dis. 2014, 70, 307–320. [Google Scholar] [CrossRef]
- Calfee, M.W.; Coleman, J.P.; Pesci, E.C. Interference with Pseudomonas Quinolone Signal Synthesis Inhibits Virulence Factor Expression by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2001, 98, 11633–11637. [Google Scholar] [CrossRef]
- Ryder, C.; Byrd, M.; Wozniak, D.J. Role of Polysaccharides in Pseudomonas aeruginosa Biofilm Development. Curr. Opin. Microbiol. 2007, 10, 644–648. [Google Scholar] [CrossRef] [Green Version]
- Jackson, K.D.; Starkey, M.; Kremer, S.; Parsek, M.R.; Wozniak, D.J. Identification of Psl, a Locus Encoding a Potential Exopolysaccharide That Is Essential for Pseudomonas aeruginosa PAO1 Biofilm Formation. J. Bacteriol. 2004, 186, 4466–4475. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.; Kolter, R. Two Genetic Loci Produce Distinct Carbohydrate-Rich Structural Components of the Pseudomonas aeruginosa Biofilm Matrix. J. Bacteriol. 2004, 186, 4457–4465. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hu, Y.; Liu, Y.; Zhang, J.; Ulstrup, J.; Molin, S. Distinct Roles of Extracellular Polymeric Substances in Pseudomonas aeruginosa Biofilm Development. Environ. Microbiol. 2011, 13, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hengzhuang, W.; Wu, H.; Damkiær, S.; Jochumsen, N.; Song, Z.; Givskov, M.; Høiby, N.; Molin, S. Polysaccharides Serve as Scaffold of Biofilms Formed by Mucoid Pseudomonas aeruginosa. FEMS Immunol. Med. Microbiol. 2012, 65, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, X.; Liu, H.; Zhang, L.; Guo, Y.; Yu, S.; Wozniak, D.J.; Ma, L.Z. The Exopolysaccharide Psl–EDNA Interaction Enables the Formation of a Biofilm Skeleton in Pseudomonas aeruginosa. Environ. Microbiol. Rep. 2015, 7, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, P.; Vallet-Gely, I.; Soscia, C.; Genin, S.; Filloux, A. The Pel Genes of the Pseudomonas aeruginosa PAK Strain Are Involved at Early and Late Stages of Biofilm Formation. Microbiology 2005, 151, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Colvin, K.M.; Gordon, V.D.; Murakami, K.; Borlee, B.R.; Wozniak, D.J.; Wong, G.C.L.; Parsek, M.R. The Pel Polysaccharide Can Serve a Structural and Protective Role in the Biofilm Matrix of Pseudomonas aeruginosa. PLoS Pathog. 2011, 7, e1001264. [Google Scholar] [CrossRef]
- Jennings, L.K.; Storek, K.M.; Ledvina, H.E.; Coulon, C.; Marmont, L.S.; Sadovskaya, I.; Secor, P.R.; Tseng, B.S.; Scian, M.; Filloux, A.; et al. Pel Is a Cationic Exopolysaccharide That Cross-Links Extracellular DNA in the Pseudomonas aeruginosa Biofilm Matrix. Proc. Natl. Acad. Sci. USA 2015, 112, 11353–11358. [Google Scholar] [CrossRef]
- Bagge, N.; Schuster, M.; Hentzer, M.; Ciofu, O.; Givskov, M.; Greenberg, E.P.; Høiby, N. Pseudomonas aeruginosa Biofilms Exposed to Imipenem Exhibit Changes in Global Gene Expression and β-Lactamase and Alginate Production. Antimicrob. Agents Chemother. 2004, 48, 1175–1187. [Google Scholar] [CrossRef]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic Resistance of Bacterial Biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Allesen-Holm, M.; Barken, K.B.; Yang, L.; Klausen, M.; Webb, J.S.; Kjelleberg, S.; Molin, S.; Givskov, M.; Tolker-Nielsen, T. A Characterization of DNA Release in Pseudomonas aeruginosa Cultures and Biofilms. Mol. Microbiol. 2006, 59, 1114–1128. [Google Scholar] [CrossRef]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Pseudomonas aeruginosa Produces an Extracellular Deoxyribonuclease That Is Required for Utilization of DNA as a Nutrient Source. Environ. Microbiol. 2010, 12, 1621–1629. [Google Scholar] [CrossRef]
- Tseng, B.S.; Zhang, W.; Harrison, J.J.; Quach, T.P.; Song, J.L.; Penterman, J.; Singh, P.K.; Chopp, D.L.; Packman, A.I.; Parsek, M.R. The Extracellular Matrix Protects Pseudomonas aeruginosa Biofilms by Limiting the Penetration of Tobramycin. Environ. Microbiol. 2013, 15, 2865–2878. [Google Scholar] [CrossRef]
- Drenkard, E.; Ausubel, F.M. Pseudomonas Biofilm Formation and Antibiotic Resistance Are Linked to Phenotypic Variation. Nature 2002, 416, 740–743. [Google Scholar] [CrossRef]
- Bass, J.I.F.; Russo, D.M.; Gabelloni, M.L.; Geffner, J.R.; Giordano, M.; Catalano, M.; Zorreguieta, Á.; Trevani, A.S. Extracellular DNA: A Major Proinflammatory Component of Pseudomonas aeruginosa Biofilms. J. Immunol. 2010, 184, 6386–6395. [Google Scholar] [CrossRef]
- O’Toole, G.A.; Kolter, R. Flagellar and Twitching Motility Are Necessary for Pseudomonas aeruginosa Biofilm Development. Mol. Microbiol. 1998, 30, 295–304. [Google Scholar] [CrossRef]
- Skerker, J.M.; Berg, H.C. Direct Observation of Extension and Retraction of Type IV Pili. Proc. Natl. Acad. Sci. USA 2001, 98, 6901–6904. [Google Scholar] [CrossRef]
- Wei, Q.; Ma, L.Z. Biofilm Matrix and Its Regulation in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2013, 14, 20983–21005. [Google Scholar] [CrossRef]
- Wagner, V.E.; Gillis, R.J.; Iglewski, B.H. Transcriptome Analysis of Quorum-Sensing Regulation and Virulence Factor Expression in Pseudomonas aeruginosa. Vaccine 2004, 22, S15–S20. [Google Scholar] [CrossRef]
- Lee, J.; Zhang, L. The Hierarchy Quorum Sensing Network in Pseudomonas aeruginosa. Protein Cell 2015, 6, 26–41. [Google Scholar] [CrossRef] [Green Version]
- Wade, D.S.; Calfee, M.W.; Rocha, E.R.; Ling, E.A.; Engstrom, E.; Coleman, J.P.; Pesci, E.C. Regulation of Pseudomonas Quinolone Signal Synthesis in Pseudomonas aeruginosa. J. Bacteriol. 2005, 187, 4372–4380. [Google Scholar] [CrossRef]
- Pamp, S.J.; Tolker-Nielsen, T. Multiple Roles of Biosurfactants in Structural Biofilm Development by Pseudomonas aeruginosa. J. Bacteriol. 2007, 189, 2531–2539. [Google Scholar] [CrossRef]
- Senturk, S.; Ulusoy, S.; Bosgelmez-Tinaz, G.; Yagci, A. Quorum Sensing and Virulence of Pseudomonas aeruginosa during Urinary Tract Infections. J. Infect. Dev. Ctries. 2012, 6, 501–507. [Google Scholar] [CrossRef]
- Jensen, V.; Löns, D.; Zaoui, C.; Bredenbruch, F.; Meissner, A.; Dieterich, G.; Münch, R.; Häussler, S. RhlR Expression in Pseudomonas aeruginosa Is Modulated by the Pseudomonas Quinolone Signal via PhoB-Dependent and -Independent Pathways. J. Bacteriol. 2006, 188, 8601–8606. [Google Scholar] [CrossRef]
- Schafhauser, J.; Lepine, F.; McKay, G.; Ahlgren, H.G.; Khakimova, M.; Nguyen, D. The Stringent Response Modulates 4-Hydroxy-2-Alkylquinoline Biosynthesis and Quorum-Sensing Hierarchy in Pseudomonas aeruginosa. J. Bacteriol. 2014, 196, 1641–1650. [Google Scholar] [CrossRef]
- Schuster, M.; Peter Greenberg, E. A Network of Networks: Quorum-Sensing Gene Regulation in Pseudomonas aeruginosa. Int. J. Med. Microbiol. 2006, 296, 73–81. [Google Scholar] [CrossRef]
- Oglesby, A.G.; Farrow, J.M.; Lee, J.-H.; Tomaras, A.P.; Greenberg, E.P.; Pesci, E.C.; Vasil, M.L. The Influence of Iron on Pseudomonas aeruginosa Physiology: A Regulatory Link Between Iron and Quorum Sensing*. J. Biol. Chem. 2008, 283, 15558–15567. [Google Scholar] [CrossRef]
- Nadell, C.D.; Drescher, K.; Wingreen, N.S.; Bassler, B.L. Extracellular Matrix Structure Governs Invasion Resistance in Bacterial Biofilms. ISME J. 2015, 9, 1700–1709. [Google Scholar] [CrossRef]
- Fleming, D.; Niese, B.; Redman, W.; Vanderpool, E.; Gordon, V.; Rumbaugh, K.P. Contribution of Pseudomonas aeruginosa Exopolysaccharides Pel and Psl to Wound Infections. Front. Cell. Infect. Microbiol. 2022, 12, e835754. [Google Scholar] [CrossRef]
- Anderl, J.N.; Franklin, M.J.; Stewart, P.S. Role of Antibiotic Penetration Limitation in Klebsiella pneumoniae Biofilm Resistance to Ampicillin and Ciprofloxacin. Antimicrob. Agents Chemother. 2000, 44, 1818–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagge, N.; Hentzer, M.; Andersen, J.B.; Ciofu, O.; Givskov, M.; Høiby, N. Dynamics and Spatial Distribution of β-Lactamase Expression in Pseudomonas aeruginosa Biofilms. Antimicrob. Agents Chemother. 2004, 48, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA Chelates Cations and Induces Antibiotic Resistance in Pseudomonas aeruginosa Biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [PubMed]
- Lewenza, S. Extracellular DNA-Induced Antimicrobial Peptide Resistance Mechanisms in Pseudomonas aeruginosa. Front. Microbiol. 2013, 4, 21. [Google Scholar] [CrossRef]
- Johnson, L.; Mulcahy, H.; Kanevets, U.; Shi, Y.; Lewenza, S. Surface-Localized Spermidine Protects the Pseudomonas aeruginosa Outer Membrane from Antibiotic Treatment and Oxidative Stress. J. Bacteriol. 2012, 194, 813–826. [Google Scholar] [CrossRef]
- Keren, I.; Mulcahy, L.R.; Lewis, K. Chapter Nineteen—Persister Eradication: Lessons from the World of Natural Products. In Methods in Enzymology; Natural Product Biosynthesis by Microorganisms and Plants, Part C.; Hopwood, D.A., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 517, pp. 387–406. [Google Scholar]
- Dörr, T.; Vulić, M.; Lewis, K. Ciprofloxacin Causes Persister Formation by Inducing the TisB Toxin in Escherichia coli. PLoS Biol. 2010, 8, e1000317. [Google Scholar] [CrossRef]
- Nguyen, D.; Joshi-Datar, A.; Lepine, F.; Bauerle, E.; Olakanmi, O.; Beer, K.; McKay, G.; Siehnel, R.; Schafhauser, J.; Wang, Y.; et al. Active Starvation Responses Mediate Antibiotic Tolerance in Biofilms and Nutrient-Limited Bacteria. Science 2011, 334, 982–986. [Google Scholar] [CrossRef]
- Zheng, Z.; Stewart, P.S. Growth Limitation of Staphylococcus Epidermidis in Biofilms Contributes to Rifampin Tolerance. Biofilms 2004, 1, 31–35. [Google Scholar] [CrossRef]
- Pagès, J.-M.; James, C.E.; Winterhalter, M. The Porin and the Permeating Antibiotic: A Selective Diffusion Barrier in Gram-Negative Bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903. [Google Scholar] [CrossRef]
- Gillis, R.J.; White, K.G.; Choi, K.-H.; Wagner, V.E.; Schweizer, H.P.; Iglewski, B.H. Molecular Basis of Azithromycin-Resistant Pseudomonas aeruginosa Biofilms. Antimicrob. Agents Chemother. 2005, 49, 3858–3867. [Google Scholar] [CrossRef]
- Wilton, M.; Charron-Mazenod, L.; Moore, R.; Lewenza, S. Extracellular DNA Acidifies Biofilms and Induces Aminoglycoside Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2015, 60, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.S.; Hennigan, R.F.; Hilliard, G.M.; Ochsner, U.A.; Parvatiyar, K.; Kamani, M.C.; Allen, H.L.; DeKievit, T.R.; Gardner, P.R.; Schwab, U.; et al. Pseudomonas aeruginosa Anaerobic Respiration in Biofilms: Relationships to Cystic Fibrosis Pathogenesis. Dev. Cell 2002, 3, 593–603. [Google Scholar] [CrossRef]
- Tolker-Nielsen, T. Pseudomonas aeruginosa Biofilm Infections: From Molecular Biofilm Biology to New Treatment Possibilities. APMIS 2014, 122, 1–51. [Google Scholar] [CrossRef]
- Burmølle, M.; Thomsen, T.R.; Fazli, M.; Dige, I.; Christensen, L.; Homøe, P.; Tvede, M.; Nyvad, B.; Tolker-Nielsen, T.; Givskov, M.; et al. Biofilms in Chronic Infections—A Matter of Opportunity—Monospecies Biofilms in Multispecies Infections. FEMS Immunol. Med. Microbiol. 2010, 59, 324–336. [Google Scholar] [CrossRef]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in Wound Repair: Molecular and Cellular Mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef]
- Pastar, I.; Nusbaum, A.G.; Gil, J.; Patel, S.B.; Chen, J.; Valdes, J.; Stojadinovic, O.; Plano, L.R.; Tomic-Canic, M.; Davis, S.C. Interactions of Methicillin Resistant Staphylococcus aureus USA300 and Pseudomonas aeruginosa in Polymicrobial Wound Infection. PLoS ONE 2013, 8, e56846. [Google Scholar] [CrossRef]
- Banu, A.; Noorul Hassan, M.M.; Rajkumar, J.; Srinivasa, S. Spectrum of Bacteria Associated with Diabetic Foot Ulcer and Biofilm Formation: A Prospective Study. Australas. Med. J. 2015, 8, 280–285. [Google Scholar] [CrossRef]
- Terreni, M.; Taccani, M.; Pregnolato, M. New Antibiotics for Multidrug-Resistant Bacterial Strains: Latest Research Developments and Future Perspectives. Molecules 2021, 26, 2671. [Google Scholar] [CrossRef]
- Melo, M.N.; Ferre, R.; Castanho, M.A.R.B. Antimicrobial Peptides: Linking Partition, Activity and High Membrane-Bound Concentrations. Nat. Rev. Microbiol. 2009, 7, 245–250. [Google Scholar] [CrossRef]
- Ahmed, T.A.E.; Hammami, R. Recent Insights into Structure–Function Relationships of Antimicrobial Peptides. J. Food Biochem. 2019, 43, e12546. [Google Scholar] [CrossRef]
- Alfei, S.; Schito, A.M. Positively Charged Polymers as Promising Devices against Multidrug Resistant Gram-Negative Bacteria: A Review. Polymers 2020, 12, 1195. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Harrison, S.D. Cell-Penetrating Peptides in Drug Development: Enabling Intracellular Targets. Biochem. Soc. Trans. 2007, 35, 821–825. [Google Scholar] [CrossRef]
- Bechinger, B.; Gorr, S.-U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef]
- Jain, A.; Duvvuri, L.S.; Farah, S.; Beyth, N.; Domb, A.J.; Khan, W. Antimicrobial Polymers. Adv. Healthc. Mater. 2014, 3, 1969–1985. [Google Scholar] [CrossRef]
- Mintzer, M.A.; Dane, E.L.; O’Toole, G.A.; Grinstaff, M.W. Exploiting Dendrimer Multivalency to Combat Emerging and Re-Emerging Infectious Diseases. Mol. Pharm. 2012, 9, 342–354. [Google Scholar] [CrossRef]
- Bahar, A.A.; Liu, Z.; Totsingan, F.; Buitrago, C.; Kallenbach, N.; Ren, D. Synthetic Dendrimeric Peptide Active against Biofilm and Persister Cells of Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 2015, 99, 8125–8135. [Google Scholar] [CrossRef]
- Gibney, K.A.; Sovadinova, I.; Lopez, A.I.; Urban, M.; Ridgway, Z.; Caputo, G.A.; Kuroda, K. Poly(ethylene imine)s as antimicrobial agents with selective activity. Macromol. Biosci. 2012, 12, 1279–1289. [Google Scholar] [CrossRef]
- Stenström, P.; Hjorth, E.; Zhang, Y.; Andrén, O.C.J.; Guette-Marquet, S.; Schultzberg, M.; Malkoch, M. Synthesis and In Vitro Evaluation of Monodisperse Amino-Functional Polyester Dendrimers with Rapid Degradability and Antibacterial Properties. Biomacromolecules 2017, 18, 4323–4330. [Google Scholar] [CrossRef] [PubMed]
- Schito, A.M.; Piatti, G.; Caviglia, D.; Zuccari, G.; Zorzoli, A.; Marimpietri, D.; Alfei, S. Bactericidal Activity of Non-Cytotoxic Cationic Nanoparticles against Clinically and Environmentally Relevant Pseudomonas Spp. Isolates. Pharmaceutics 2021, 13, 1411. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.M.V.; Crusz, S.A.; Kolomiets, E.; Buts, L.; Kadam, R.U.; Cacciarini, M.; Bartels, K.-M.; Diggle, S.P.; Cámara, M.; Williams, P.; et al. Inhibition and Dispersion of Pseudomonas aeruginosa Biofilms by Glycopeptide Dendrimers Targeting the Fucose-Specific Lectin LecB. Chem. Biol. 2008, 15, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Kolomiets, E.; Swiderska, M.A.; Kadam, R.U.; Johansson, E.M.V.; Jaeger, K.-E.; Darbre, T.; Reymond, J.-L. Glycopeptide Dendrimers with High Affinity for the Fucose-Binding Lectin LecB from Pseudomonas aeruginosa. ChemMedChem 2009, 4, 562–569. [Google Scholar] [CrossRef]
- Hou, S.; Zhou, C.; Liu, Z.; Young, A.W.; Shi, Z.; Ren, D.; Kallenbach, N.R. Antimicrobial dendrimer active against Escherichia coli biofilms. Bioorg. Med. Chem. Lett. 2009, 19, 5478–5481. [Google Scholar] [CrossRef]
- Johansson, E.M.; Kadam, R.U.; Rispoli, G.; Crusz, S.A.; Bartels, K.-M.; Diggle, S.P.; Cámara, M.; Williams, P.; Jaeger, K.-E.; Darbre, T.; et al. Inhibition of Pseudomonas aeruginosa Biofilms with a Glycopeptide Dendrimer Containing D-Amino Acids. MedChemComm 2011, 2, 418–420. [Google Scholar] [CrossRef]
- Kadam, R.U.; Bergmann, M.; Hurley, M.; Garg, D.; Cacciarini, M.; Swiderska, M.A.; Nativi, C.; Sattler, M.; Smyth, A.R.; Williams, P.; et al. A Glycopeptide Dendrimer Inhibitor of the Galactose-Specific Lectin LecA and of Pseudomonas aeruginosa Biofilms. Angew. Chem. Int. Ed. 2011, 50, 10631–10635. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Zhou, C.; Kallenbach, N.R.; Ren, D. Control of Bacterial Persister Cells by Trp/Arg-Containing Antimicrobial Peptides. Appl. Environ. Microbiol. 2011, 77, 4878–4885. [Google Scholar] [CrossRef]
- Wang, L.; Erasquin, U.J.; Zhao, M.; Ren, L.; Zhang, M.Y.; Cheng, G.J.; Wang, Y.; Cai, C. Stability, Antimicrobial Activity, and Cytotoxicity of Poly(Amidoamine) Dendrimers on Titanium Substrates. ACS Appl. Mater. Interfaces 2011, 3, 2885–2894. [Google Scholar] [CrossRef]
- Scorciapino, M.A.; Pirri, G.; Vargiu, A.V.; Ruggerone, P.; Giuliani, A.; Casu, M.; Buerck, J.; Wadhwani, P.; Ulrich, A.S.; Rinaldi, A.C. A Novel Dendrimeric Peptide with Antimicrobial Properties: Structure-Function Analysis of SB056. Biophys. J. 2012, 102, 1039–1048. [Google Scholar] [CrossRef]
- Lu, Y.; Slomberg, D.L.; Shah, A.; Schoenfisch, M.H. Nitric Oxide-Releasing Amphiphilic Poly(Amidoamine) (PAMAM) Dendrimers as Antibacterial Agents. Biomacromolecules 2013, 14, 3589–3598. [Google Scholar] [CrossRef]
- Reymond, J.L.; Bergmann, M.; Darbre, T. Glycopeptide dendrimers as Pseudomonas aeruginosa biofilm inhibitors. Chem. Soc. Rev. 2013, 42, 4814–4822. [Google Scholar] [CrossRef]
- Zielinska, P.; Staniszewska, M.; Bondaryk, M.; Koronkiewicz, M.; Urbanczyk-Lipkowska, Z. Design and Studies of Multiple Mechanism of Anti-Candida Activity of a New Potent Trp-Rich Peptide Dendrimers. Eur. J. Med. Chem. 2015, 105, 106–119. [Google Scholar] [CrossRef]
- Worley, B.V.; Schilly, K.M.; Schoenfisch, M.H. Anti-Biofilm Efficacy of Dual-Action Nitric Oxide-Releasing Alkyl Chain Modified Poly(Amidoamine) Dendrimers. Mol. Pharm. 2015, 12, 1573–1583. [Google Scholar] [CrossRef]
- Visini, R.; Jin, X.; Bergmann, M.; Michaud, G.; Pertici, F.; Fu, O.; Pukin, A.; Branson, T.R.; Thies-Weesie, D.M.E.; Kemmink, J.; et al. Structural Insight into Multivalent Galactoside Binding to Pseudomonas aeruginosa Lectin LecA. ACS Chem. Biol. 2015, 10, 2455–2462. [Google Scholar] [CrossRef]
- Backlund, C.J.; Worley, B.V.; Schoenfisch, M.H. Anti-Biofilm Action of Nitric Oxide-Releasing Alkyl-Modified Poly(Amidoamine) Dendrimers against Streptococcus Mutans. Acta Biomater. 2016, 29, 198–205. [Google Scholar] [CrossRef]
- Bergmann, M.; Michaud, G.; Visini, R.; Jin, X.; Gillon, E.; Stocker, A.; Imberty, A.; Darbre, T.; Reymond, J.-L. Multivalency Effects on Pseudomonas aeruginosa Biofilm Inhibition and Dispersal by Glycopeptide Dendrimers Targeting Lectin LecA. Org. Biomol. Chem. 2016, 14, 138–148. [Google Scholar] [CrossRef]
- Michaud, G.; Visini, R.; Bergmann, M.; Salerno, G.; Bosco, R.; Gillon, E.; Richichi, B.; Nativi, C.; Imberty, A.; Stocker, A.; et al. Overcoming Antibiotic Resistance in Pseudomonas aeruginosa Biofilms Using Glycopeptide Dendrimers. Chem. Sci. 2016, 7, 166–182. [Google Scholar] [CrossRef]
- Batoni, G.; Casu, M.; Giuliani, A.; Luca, V.; Maisetta, G.; Mangoni, M.L.; Manzo, G.; Pintus, M.; Pirri, G.; Rinaldi, A.C.; et al. Rational Modification of a Dendrimeric Peptide with Antimicrobial Activity: Consequences on Membrane-Binding and Biological Properties. Amino Acids 2016, 48, 887–900. [Google Scholar] [CrossRef]
- Vankoten, H.W.; Dlakic, W.M.; Engel, R.; Cloninger, M.J. Synthesis and Biological Activity of Highly Cationic Dendrimer Antibiotics. Mol. Pharm. 2016, 13, 3827–3834. [Google Scholar] [CrossRef]
- Barrios-Gumiel, A.; Sanchez-Nieves, J.; Pérez-Serrano, J.; Gómez, R.; de la Mata, F.J. PEGylated AgNP Covered with Cationic Carbosilane Dendrons to Enhance Antibacterial and Inhibition of Biofilm Properties. Int. J. Pharm. 2019, 569, 118591. [Google Scholar] [CrossRef]
- Gómez-Casanova, N.; Lozano-Cruz, T.; Soliveri, J.; Gomez, R.; Ortega, P.; Copa-Patiño, J.L.; Heredero-Bermejo, I. Eradication of Candida albicans Biofilm Viability: In Vitro Combination Therapy of Cationic Carbosilane Dendrons Derived from 4-Phenylbutyric Acid with AgNO3 and EDTA. J. Fungi 2021, 7, 574. [Google Scholar] [CrossRef]
- Fernandez, J.; Martin-Serrano, Á.; Gómez-Casanova, N.; Falanga, A.; Galdiero, S.; Javier de la Mata, F.; Heredero-Bermejo, I.; Ortega, P. Effect of the Combination of Levofloxacin with Cationic Carbosilane Dendron and Peptide in the Prevention and Treatment of Staphylococcus aureus Biofilms. Polymers 2021, 13, 2127. [Google Scholar] [CrossRef]
- Galdiero, E.; de Alteriis, E.; De Natale, A.; D’Alterio, A.; Siciliano, A.; Guida, M.; Lombardi, L.; Falanga, A.; Galdiero, S. Eradication of Candida albicans Persister Cell Biofilm by the Membranotropic Peptide GH625. Sci. Rep. 2020, 10, 5780. [Google Scholar] [CrossRef] [Green Version]
- Quintana-Sanchez, S.; Gómez-Casanova, N.; Sánchez-Nieves, J.; Gómez, R.; Rachuna, J.; Wasik, S.; Semaniak, J.; Maciejewska, B.; Drulis-Kawa, Z.; Ciepluch, K.; et al. The Antibacterial Effect of PEGylated Carbosilane Dendrimers on P. Aeruginosa Alone and in Combination with Phage-Derived Endolysin. Int. J. Mol. Sci. 2022, 23, 1873. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Bacterium | Drug(s) Resistant to | Typical Disease |
|---|---|---|
| E. coli | Cephalosporins, fluoroquinolones | UTI, BSI |
| K. pneumoniae | Cephalosporins, carbapenems | Pneumonia, BSI, UTI |
| S. aureus | Methicillin | Wound, BSI |
| S. pneumoniae | Penicillin | Pneumonia, meningitis, otitis |
| Nontyphoidal Salmonella | Fluoroquinolones | Foodborne diarrhea, BSI |
| Shigella spp. | Fluoroquinolones | Diarrhea * |
| N. gonorrhoeae | Cephalosporins | Gonorrhea |
| M. tuberculosis | Rifampicin, isoniazid, fluoroquinolone | Tuberculosis |
| Name of Fungi | ||
| Candida spp. | Fluconazole, echinocandins [7] | Candidiasis |
| Cryptococcus neoformans | Fluconazole [8] | Cryptococcosis |
| Aspergillus spp. | Azoles [9] | Aspergillosis |
| Scopulariopsis spp. Onychomycosis | Amphotericin B, flucytosine, azoles [10] | Onychomycosis |
| Name of Virus | ||
| Cytomegalovirus (CMV) | Ganciclovir, foscarnet [11] | AIDS and oncology patients |
| Herpes simplex virus (HSV) | Acyclovir, famciclovir, valacyclovir [12] | Herpes simplex |
| Human immunodeficiency virus (HIV) | Antiretroviral drugs [13] | AIDS |
| Influenza virus | Amantadine, rimantadine, neuraminidase inhibitors [14] | Influenza |
| Varicella zoster virus | Acyclovir, valacyclovir [12] | Chicken pox |
| Hepatitis B virus (HBV) | Lamivudine [15] | Hepatitis B |
| Name of Parasite | ||
| Plasmodia spp. | Chloroquine, artemisinin, atovaquone [16] | Malaria |
| Leishmania spp. | Pentavalent antimonials, miltefosine paromomycin, amphotericin B [17,18] | Leishmaniasis |
| Schistosomes | Praziquantel, oxamniquine [19,20] | Schistosomiasis |
| Entamoeba | Metronidazole [21] | Amoebiasis |
| Trichomonas vaginalis | Nitroimidazoles [22] | Trichomoniasis |
| Toxoplasma gondii | Artemisinin, atovaquone, sulfadiazine [23,24,25] | Toxoplasmosis |
| BF Biomass | Organized communities of pathogens | Sessile cells | Cells attached to surfaces forming highly coordinated microcolonies, communicating by the QS system, and lacking motility |
| Persistent cells | Small subpopulation of microorganisms reversibly transformed into slowly growing cells | ||
| Dormant cells | |||
| Extracellular polymeric substances (EPSs) | Enzymatic proteins | Polypeptides | |
| Polysaccharides | Cellulose Polyglucosamine (PGA) Anexopolysaccharides | ||
| Extracellular DNA (eDNA) | |||
| Cationic and anionic glycoproteins | |||
| Cationic and anionic glycolipids | Allow real communication between bacteria Stabilize the 3-D structure of BF | ||
| Functions | Components | Sub Components | Molecules | Function |
|---|---|---|---|---|
| Matrix Adhesive material Protective barrier | ECMs | EPS | PsL * | Initiation and maintenance of BF Supplies cell surface attachment Supplies intercellular interactions |
| PeL *, ** | Essential for forming pellicles at the air–liquid interface and solid surface-associated BFs [98,102] Platform for BF structure Supplies protection against aminoglycosides [100,103] Binding with eDNA of the BF [104,105]. Compensates for a lack of PsL in the BF periphery [104] | |||
| Alginate *** | Factor used to distinguish mucoid or non-mucoid P. aeruginosa BFs Retains water and nutrients Supplies antibiotic resistance and immune evasion [105,106,107] | |||
| eDNA | Formation of cation gradients Antibiotic resistance Nutrient source Early BF development [99,108,109,110] Major proinflammatory factor for P. aeruginosa BFs [111] | |||
| Proteins | Flagella | Act as an adhesin to help initial bacterial attachment to the surface [112] | ||
| Type IV pili | Contribute to the formation of mushroom-like BF cap structures [113,114] | |||
| CdrA adhesin | Interacts with PsL and increases BF stability [115]. | |||
| Cup fimbriae | Important roles in cell-to-cell interaction during the first stage of BF formation [115] | |||
| Intercellular communication system enabling bacteria to sense their own population density. | QS system | NAHSL AI-2 | las | Regulate several hundred genes in P. aeruginosa [116,117] Regulate the bacterial phenotype, spatial differentiation in BFs, motility, and BF formation [118] |
| Rhl | ||||
| PQS | ||||
| Integrated QS (IQS) | ||||
| Reasons for the Failure of Antibiotics | BF Function | Factors | Inactivated Antibiotics | Ref. |
|---|---|---|---|---|
| Hampered antibiotic penetration | Anti-spread barrier | EPS | Ampicillin Ciprofloxacin | [126] |
| Presence of antibiotic-degrading enzymes | To provide β-lactamases (β-LS) | ↑ β-LS | Ampicillin | [127] |
| Imipenem Ceftazidime | ||||
| Increased BF resistance | To provide eDNA | ↑ eDNA ↓ Mg2+ | Cationic Peptides Aminoglycosides | [128,129,130] |
| Presence of persistent cells | To cause gradients in nutrients and oxygen concentration To promote differentiation in cell growth | Endogenous stress TA 1-systems | Rifampicin Aminoglycosides | [131] |
| Presence of dormant cells | ↓ Functions ↓ Energy ↓ Biosynthesis | Fluoroquinolones | [132] | |
| ↑ Resistance to stress | To cause adaptive stress responses by heterogeneity | Changes in component/processes target of antibiotics | Ofloxacin Gentamicin Meropenem Colistin | [133] |
| Ofloxacin | [134] | |||
| ↑ Export of membrane proteins | To up-regulate the production of some efflux pumps | ↑ Efflux pumps QS | Multidrugs | [135] |
| Azithromycin | [136] | |||
| Genetic diversity | To act as a reservoir of genetic diversity by promoting plasmids transfer | Horizontal gene transfer (HGT) eDNA QS | Aminoglycosides | [137] |
| Dendrimers Structure | Name | Activity |
|---|---|---|
 | 2G3 | IC50 = 0.025 µM P. aeruginosa (PA) LecB |
 | (RW)4D | Inactivate E. coli RP437 planktonic culture and BFs |
 | FD2 | LD50 LecB PA 0.14 µM P. aeruginosa BF formation (IC50 = 10 mM) |
| D-FD2 | LD50 LecB PA 0.66 µM | |
 | PEGylated PAMAM Film on MAO Substrate | BF by PA (strain PAO1) and S. aureus (SA). |
 | Nitric Oxide-Releasing amphiphilic PAMAM | Inhibited PA BFs |
 | 14 | Inhibited C. albicans BF |
 | G4 PAMAMs decorated with C16-DABCO | Inhibited E. coli and B. cereus BF |
 | PEGylated AgNPs covered with cationic carbosilane dendrons | Inhibited E. coli and S. aureus BF |
| Mechanism of Action | Type of Dendrimers | Target Pathogens | Preventing BF Formation | Hindering BF Development | BF Dispersal | Joint Therapy | [Ref.] Year |
|---|---|---|---|---|---|---|---|
| Inhibition LecB | FD2 | P. aeruginosa |  | |  | | [156] 2008 |
| 2G3 | | | | | [157] 2009 | ||
| Membrane (Ms) Disruptors | (RW) 4D | E. coli | | | | | [158] 2009 |
| Inhibition LecB | D-FD2 | P. aeruginosa | | | | | [159] 2011 |
| Inhibition LecA | GalAG2 and GalBG2 | | | | | [160] 2011 | |
| Ms disruptors | (RW)4-NH2 | E. coli | | | | 20 µM +0.5 µg/mL * 0% BF cells | [161] 2011 |
| (RW) 4D | E. coli HM22 | | | | 20 µM +0.5 µg/mL * <10% BF cells | ||
| Ti-S(CaPO4)060-PEGPAMAMs Ti-S-060-PEGPAMAMs | P. aeruginosa S. aureus | | | | | [162] 2011 | |
| SB056 | S. epidermidis P. aeruginosa | | | | | [163] 2012 | |
| Ms disruptors Release of NO | NO-releasing PAMAMs # | P. aeruginosa | | | | | [164] 2013 |
| Inhibition LecA Inhibition LecB | TGPDs | P. aeruginosa | | | | | [165] 2013 |
| Membranolytic Apoptotic | Cationic antifungal peptide dendrimers (9, 14) | C. albicans | | | | | [166] 2015 |
| Outer cell wall damage Absence of true hyphae Filamentation inhibition Ms permeabilization | AgNPs | C. albicans | | | | | [59] 2015 |
| Outer cell wall damage Ms permeabilization BF mass penetration | 2D-24 | P. aeruginosa | | | |
Ciprofloxacin (Cip) 1 Tobramycin (Tob) **, 1 Carbenicillin (Car) 1 | [152] 2015 |
| Ms disruptors Release of NO | (G1–G4)-NO-releasing alkyl-PAMAMs # | P. aeruginosa S. aureus MRSA | | | | | [167] 2015 |
| Electrostatic interactions Ms disruptors Release of NO |
(G1)-NO-releasing alkyl PAMAMs # | S. mutans | | | | | [169] 2016 |
| Inhibition LecA | G3/G4 analogous of GalAG2 and GalBG2 | P. aeruginosa | | | | | [170] 2016 |
| Inhibition LecA Inhibition LecB | Het1G2-Het8G2 Het1G1Cys-Het8G1Cys Others | P. aeruginosa | | | | Dendrimer FD2+Tob | [171] 2016 |
| Ms disruptors | den-SB056-1 | S. epidermidis |  | | | | [172] 2016 |
| C16-DABCO-G4-PAMAM |
S. aureus
B. cereus |  § § | | | | [173] 2016 | |
| NH4+ carbosilane dendrimers and dendrons (CBSD) | S. aureus CECT240 | | | | | [78] 2016 | |
| Ms disruptor Ag release | PEGylated-NH4+ CBSD-AgNPs |
S. aureus
E. coli | | | | | [174] 2019 |
| Ms disruptor | BDSQ024 | C. albicans | | | |
Amphotericin (AmphB) Caspofungin (CSF) | [60] 2020 |
| ArCO2G2 (SNMe3I)4 | C. albicans | | | 2 | Ethylenediaminetetraacetic acid (EDTA) + silver nitrate (AgNO3) | [175] 2021 | |
| Interaction/disruption of the Ms bilayers | MalG2(SNHMe 2Cl)4 DPC | S. aureus | 2 | 2 | | Levofloxacin (LEV) | [176] 2021 |
| Interaction/disruption of the Ms bilayers No pore formation | gH625-M 3 | C. tropicalis C. serratia C. marcescens S. aureus | | | | AmphB 4 Fluconazole (FC) Echinocandins 4 5 Flucytosine (5-FLC) | [177] 2020 |
| Interaction/disruption of the Ms bilayers | PEG/no-PEG cationic CBSD | P. aeruginosa | 2 | 2 | 2 | Endolysin | [178] 2022 |
= reduced with respect to SB056; § the treatment of membranes with 1 mg/mL causes complete inhibition of the growth of S. aureus BFs; 2 when in combination as in the column 7; 3 analogous of the cell penetratin peptide gH625; 4 not functioning; HM22 denotes a strain containing the hipA7 allele, which maps to the hipA gene in the antitoxin-toxin module HipBA. The expression of the hipA7 allele confers a 1000-fold higher frequency of persister cells formation; AgNPs = silver nanoparticles; MRSA = methicillin-resistant S. aureus; = not reported; = yes, active; = no, not active.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfei, S.; Caviglia, D. Prevention and Eradication of Biofilm by Dendrimers: A Possibility Still Little Explored. Pharmaceutics 2022, 14, 2016. https://doi.org/10.3390/pharmaceutics14102016
Alfei S, Caviglia D. Prevention and Eradication of Biofilm by Dendrimers: A Possibility Still Little Explored. Pharmaceutics. 2022; 14(10):2016. https://doi.org/10.3390/pharmaceutics14102016
Chicago/Turabian StyleAlfei, Silvana, and Debora Caviglia. 2022. "Prevention and Eradication of Biofilm by Dendrimers: A Possibility Still Little Explored" Pharmaceutics 14, no. 10: 2016. https://doi.org/10.3390/pharmaceutics14102016
APA StyleAlfei, S., & Caviglia, D. (2022). Prevention and Eradication of Biofilm by Dendrimers: A Possibility Still Little Explored. Pharmaceutics, 14(10), 2016. https://doi.org/10.3390/pharmaceutics14102016