1. Introduction
Parasitic gastroenteritis is a major challenge to small ruminant production and has been associated with substantial economic losses and welfare concerns [
1]. The abomasal hematophagous nematode
Haemonchus contortus (
H. contortus) is the most economically important parasite, responsible for a tremendous reduction in sheep productivity [
2]. Anthelmintics are routinely used to control parasitism but concerns over residues, and development of multidrug-resistant parasite strains threaten the sustainability of this approach, driving the need for an alternative and more sustainable parasite control measures [
3]. Researchers have been delving into the possibility of using bioactive phyto-chemicals as an alternative to anthelmintics, hoping to reduce the reliance on synthetic chemicals [
4].
Essential oils (EOs) and their constituents possess remarkable biological activities [
5,
6,
7]. In a previous study, the essential oil coriander and three EO components (linalool, geraniol and eugenol) were found to have a nematocidal effect against several ovine gastrointestinal nematodes [
8]. However, the use of EOs is hampered by the limited thermal and chemical stability, poor efficacy at low concentrations, and the narrow margin of safety [
9]. Encapsulation of EOs within a polymeric matrix can protect the bioactive compounds of EOs, enhance their biological properties, reduce toxicity, and facilitate a targeted and controlled delivery [
10]. Among the common biodegradable polymers, poly(lactic-co-glycolic acid) (PLGA) is widely used as a polymeric carrier in hydrophobic drug delivery, such as EO components, for production of micro- or nanoparticles [
11].
PLGA particles can be formulated by conventional methods, such as emulsification-solvent evaporation and nanoprecipitation techniques. However, these methods are not reproducible, and associated with broad size distribution and less entrapment efficiency [
12]. In contrast, microfluidic technology enables the production of monodisperse particles with varying sizes and morphological features in high-throughput and reproducible batches via controlling the fluid flow [
13]. Two microfluidic methods are often used to produce either micron-scale particles with a higher encapsulation efficiency and drug loading capacity (droplet-based flow focusing method) or nano-scale particles with a relatively low entrapment efficiency and drug loading capacity (continuous flow-focusing method) [
14]. Synthesis of particles with a specific size without compromising their entrapment and loading capacities can be achieved by a continuous flow-focusing method, using a partially water-miscible solvent mixture as the dispersed phase, which produces a consistent flow when mixing with a continuous aqueous phase, producing discrete microdroplets that lead to the formation of nano-scale particles [
15].
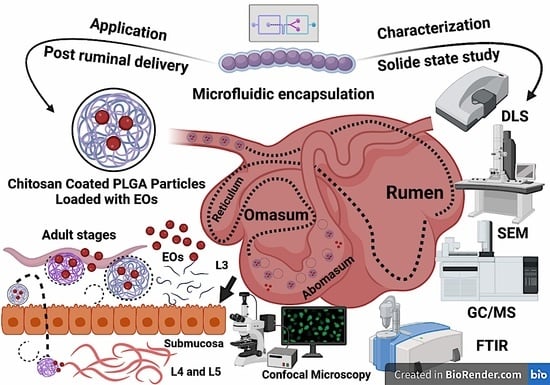
The post-ruminal (abomasal) delivery of drugs via the oral route is often hampered by mechanical disruption by rumination, chemical degradation, microbial fermentation, and ruminal retention [
16]. Coating the surface of PLGA particles with chitosan can transform anionic PLGA particles into cationic, chitosan (CS)-coated PLGA particles (CS-PLGA) [
17]. This modification improves the mucoadhesive properties of the particles, prolongs the retention time within the mucosal tissue, and sustains the release kinetics [
18]. Additionally, chitosan has a good solubility in acidic media [
19], enabling the release of EO components in the abomasum, where
H. contortus live. Chitosan can also enhance the penetration of the entrapped drug to the submucosa via opening the tight-junctions of the intestinal barrier [
20].
In the present study, we tested the hypothesis that encapsulating EO components (linalool, eugenol, and geraniol) into CS-PLGA particles improves EO component bioavailability and maintains their efficacy against gastrointestinal nematodes, particularly H. contortus. We produced micro and nano-particles of CS-PLGA encapsulating linalool, eugenol, and geraniol, individually or combined. The hydrodynamic size, polydispersity index (PDI), and ζ-potential of the CS-PLGA particles were characterized. We also investigated the entrapment efficiency, loading capacity, and in vitro release kinetic of EO components from loaded particles using gas chromatography mass spectrometry (GC-MS) and UV- vis spectrophotometer. A further solid-state characterization was performed using Fourier transform infrared (FT-IR) spectroscopy, thermogravimetric analysis (TGA), and X-ray powder diffraction (XRD). The antiparasitic activity of the particles was tested against larval and adult stages of H. contortus and the structural changes of the cuticle was examined using scanning electron microscopy (SEM). Finally, we assessed the effect of particles loaded with EO components on host cell viability, proliferation, membrane integrity, drug permeability, and cellular uptake using a bovine gut cell line (FFKD-1-R).
2. Materials and Methods
2.1. Chemicals and Reagents
The following chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA): Poly(D,L-lactide-co-glycolide) (PLGA; lactide:glycolide 65:35, MW 40–75 KDa), Poly(vinyl alcohol) (PVA; 87%–89% hydrolyzed, MW = 13–23 KDa), sucrose, sodium tripolyphosphate (TPP; MW = 367.86), medium molecular weight chitosan (MW = 50–190 KDa), polysorbate 80, span 20, fluorescein 5(6)-isothiocyanate (FITC), clove oil (Eugenia caryophyllata), lavender oil (Lavandula angustifolia), pure components (~100% purity), and analytical standards of linalool and geraniol. Pure eugenol was obtained from flourochem Ltd. (Glossop, UK). The essential oils citronella (Cymbopogon nardus) and coriander (Coriandrum sativum) were purchased from Oils4life Ltd. (Great Yarmouth, UK). Dichloromethane (DCM) and acetone were obtained from Fisher Scientific Inc. The analytical standards of eugenol were purchased from the European Pharmacopoeia (EP) reference standards.
For cell culture work, bovine intestinal cell line of jejunum/ileum (FKD-1-R; Cat No. CCLV-RIE 0971) was kindly provided by Friedrich-Loeffler-Institute, Federal Research Institute for Animal Health, Germany. Dulbecco’s Modified Eagle’s Medium—low glucose (DMEM), heat-inactivated fetal bovine serum (FBS), trypsin-EDTA Solution 10X, Dulbecco’s phosphate buffer saline (DPBS), trypan blue, and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) solution were purchased from Sigma-Aldrich. Penicillin-streptomycin (10,000 U/mL) and dimethyl sulfoxide (DMSO—cell culture grade) were purchased from Fisher Scientific—Gibco™. IncuCyte® Cytotox red dye was purchased from Sartorius, USA. Phalloidin-iFluor 555 reagent (ab176756) and DAPI (ab285390) were obtained from Abcam, Cambridge, UK.
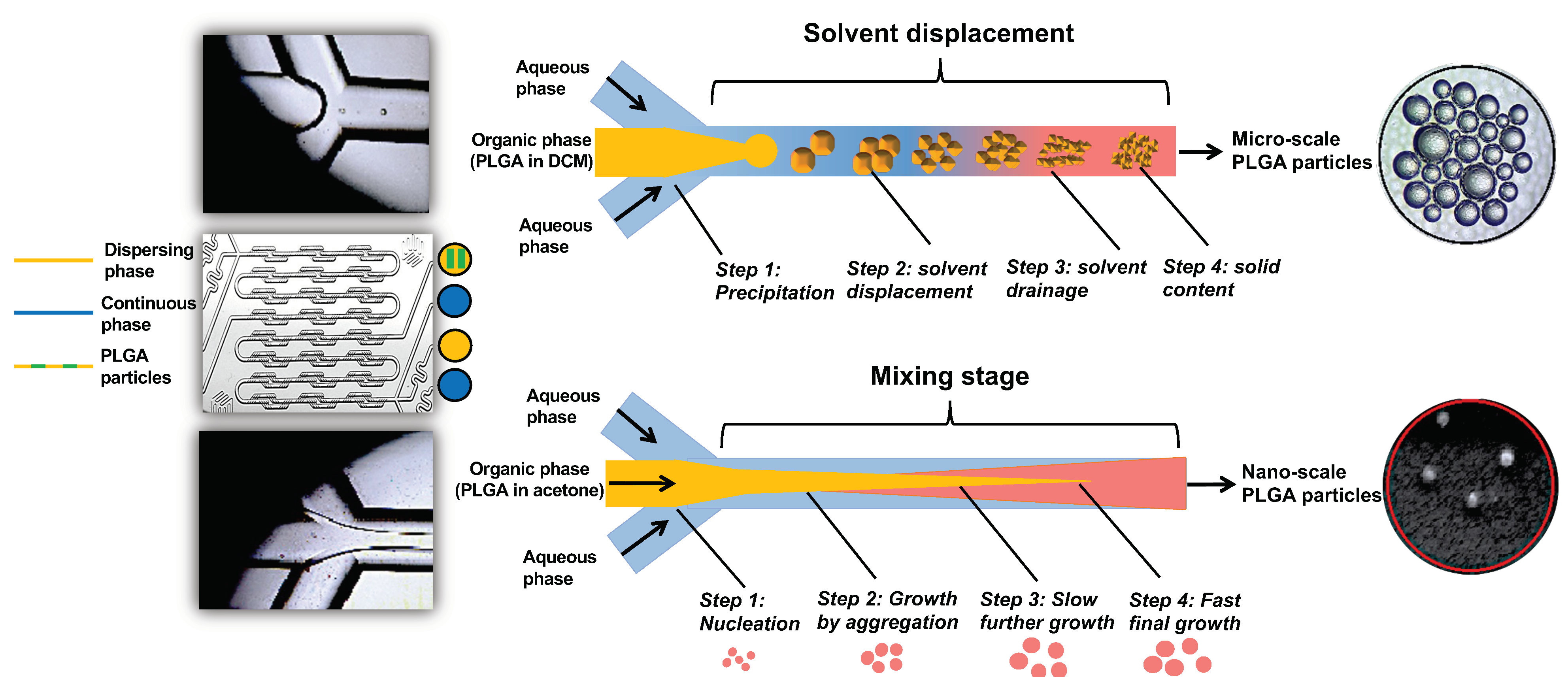
2.2. Microfluidic System and Chip
The micromixer system, developed by The Dolomite Centre Ltd., Royston, UK, was used to prepare PLGA particles at micro and nanoscales. The system uses a microfluidic hydrodynamic focusing (MHF) technique and consists of a micromixer chip (Dolomite, 3200401), a hydrophilic compact glass microfluidic device that enables rapid mixing of two fluids with 12 herring bone mixing stages to produce the particles in a controlled and reproducible way. The chip was assembled with the H Interface (Dolomite, Part No. 3000155) and two Linear Connectors 4-way (Dolomite, Part No. 3000024). The fluids were delivered by two pressure pumps (Fluika) through FEP Tubing, 1/16 × 0.25 mm and the stream was controlled by 2-way in-line valves and visualized via a high-speed digital microscope. The process of particle formulation using a micromixer chip by solvent displacement (acetone) or solvent diffusion (DCM) is illustrated in
Figure 1.
2.3. Chemical Analysis
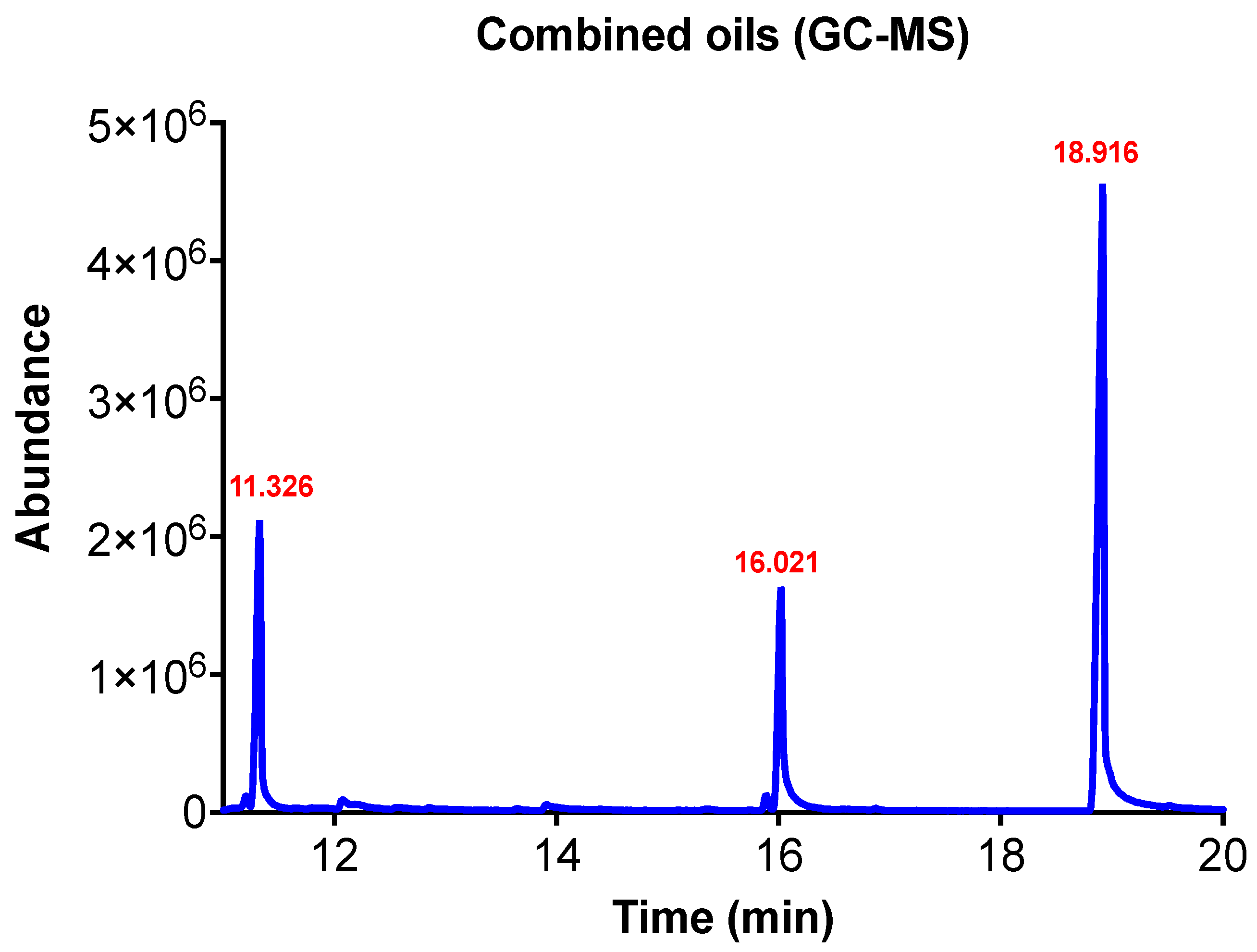
The chemical composition of EOs citronella, clove, coriander, and lavender was determined by qualitative and quantitative methods. The headspace (Turbo-Matrix HS 16 sampler, PerkinElmer, Waltham, MA, USA) coupled with gas chromatography-mass spectrometry (Clarus
® SQ 8 GC/MS PerkinElmer, USA) was used to identify the volatile components of each EO [
21]. The quantitative analysis of EOs was carried out on gas chromatography (Agilent 6890N GC system) equipped with a flame ionization detector (FID) and HP-5MS capillary column (30 m × 0.25 mm i.d., 0.25 µm film thickness) as described previously [
22] with minor modifications. Briefly, 1 µL of 1% EO diluted in methanol was injected in splitless mood. The injector temperature was set at 250 °C. Helium was used as the carrier gas flowing at 1 mL/min and pressure of 7.6 bar. The initial temperature of the column was 50 °C for 2 min and increased to 200 °C at a rate of 5 °C/min and maintained for 3 min at the final temperature. The electron impact mass spectra were scanned at 70 ev in the mass range of 40–600 units. Each sample was analyzed in triplicate with a total run time of 37 min. The chemical components were detected via matching their mass spectra and retention times to the NIST library data. The peak area of each EO component was compared to a calibration curve constructed from the values of five known concentrations of a reference standard of the respective EO component.
2.4. Preparation of PLGA and CS-PLGA Particles
To synthesize PLGA particles, PLGA 1 and 2% (w/v) was dissolved in organic solvent (organic phase) and rapidly mixed with water (aqueous phase) in the Dolomite micromixer chip to formulate the oil-in-water (O/W) emulsion. The organic solvents used were dichloromethane (DCM) and acetone either individually or in combinations (1:10 and 1:20, respectively) to optimize the formulation condition. Due to the hydrophobic nature of the EOs, the EO components (geraniol, linalool, and eugenol) were dispersed in the organic phase with different concentrations (0.25, 0.5, and 1%). The working concentrations were determined based on the particle size and entrapment efficiency percentage (EE%) of the EO components chosen for chitosan coating.
The particles were prepared via microfluidic hydrodynamic focusing as previously described [
14], with some modifications. Based on the preliminary results of optimization, 100 mg PLGA was dissolved in 10 mL solvent (DCM: acetone mixture; 1:10) to form the organic phase. The aqueous phase was prepared by dissolution PVA 1% (
w/
v) in deionized water. The two fluids were mixed in the dolomite’s micromixer system at a flow rate 1000 µL/min with an operating pressure 500:250 mbar for dispersing to a continuous phase. For encapsulation, the EO components were mixed individually or in combination with the organic phase 0.5% (
v/
v). The hydrophilic lipophilic balance (HLB) was adjusted by addition of span 80 to the organic phase (1%
v/
v) and tween 80 to the aqueous phase (1%
v/
v) to help emulsification of oils in distilled water. To modify the PLGA particles surface, chitosan was dissolved in 1% glacial acetic acid (0.25%), stirred overnight, and mixed with the aqueous phase. The PLGA particles were collected from the chip’s output to a preloaded reservoir with 10 mL of 0.1% PVA solution. For cross-linking of chitosan, 0.4% of TPP solution was gradually added to the emulsion with continuous stirring to form ionic gelation. The organic solvent was then evaporated overnight under a moderate magnetic stirring and the particles were recovered by centrifugation at 10,000 rpm for 30 min at 4 °C and washed three times with distilled water. The pellet was suspended in distilled water to form a homogenous suspension and subsequently lyophilized in a freeze dryer at −65 °C for 72 h (Labconco, Eaton Socon, UK).
2.5. Characterization of the Prepared Particles
2.5.1. Photon Correlation Spectroscopy and Zeta-Potential Analysis
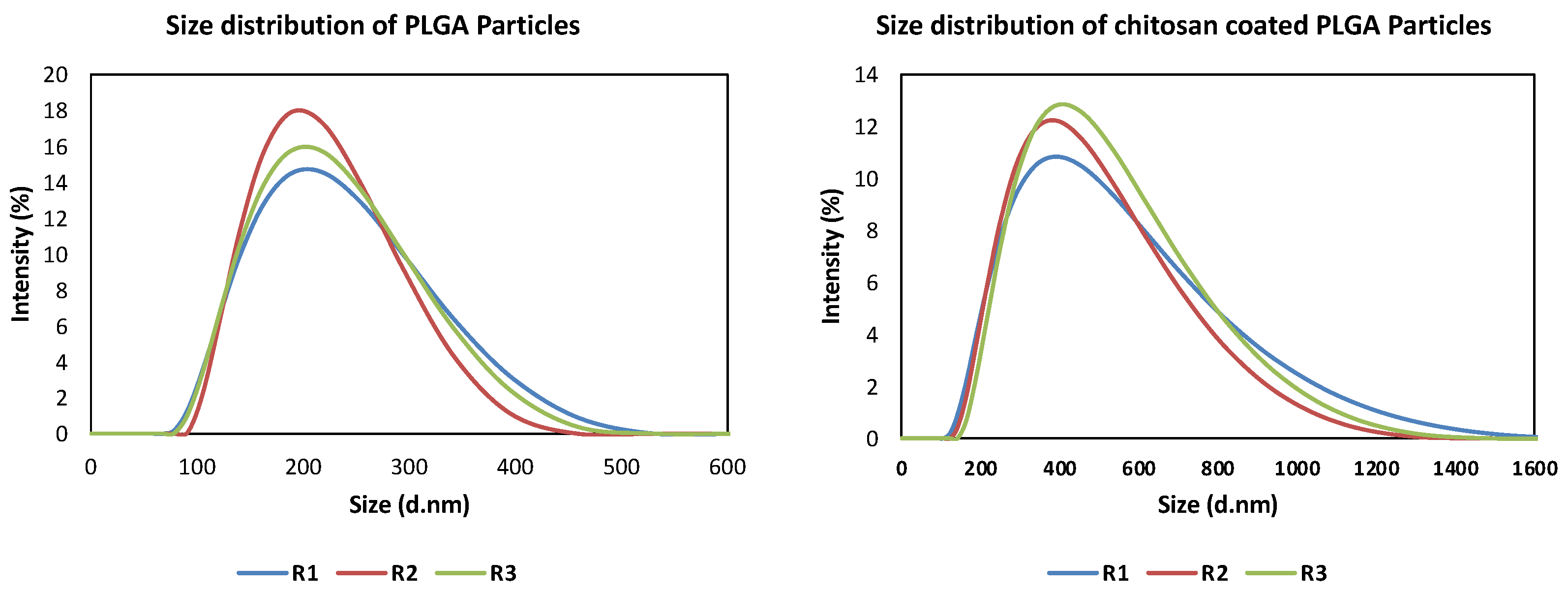
The size, polydispersity index (PDI), distribution, and homogeneity of the prepared particles were examined using dynamic light scattering (DLS) and photon correlation spectroscopy (PCS), while the Zeta (ζ)-potential was determined using a Zetasizer Nanoseries ZS (Malvern instruments, Malvern, UK). Freshly prepared and lyophilized particles were diluted with distilled water to a final concentration of 1 mg/mL to obtain evenly distributed particles. The measurement was performed at room temperature with a refractive index of 1.33 and a viscosity of 0.8872 cp. Each sample was analyzed in triplicate and the results are presented as the means ± standard deviations (S.D.).
2.5.2. Entrapment Efficiency (EE%) and Drug Loading Capacity (LC%)
The encapsulation efficiency and EO components content of the prepared particles were determined by GC/MS chromatography as described previously [
7], with some modifications. Briefly, 5 mg of the lyophilized sample was dissolved in 1 mL methanol to degrade the polymer and release the EO components. This step was enhanced by subsequent sonication–vortexing cycles. To precipitate the polymer, the samples were centrifuged at 13,000 rpm for 15 min and the concentration of EO components was quantified in the clear supernatant using a calibration curve constructed by using five known concentrations of a reference standard. The plain particles were treated using the same procedure and used as a blank. Triplicate of each prepared particle were analyzed. The EE% and LC% were calculated according to the following formulas [
20]:
2.5.3. Morphology of the Particles
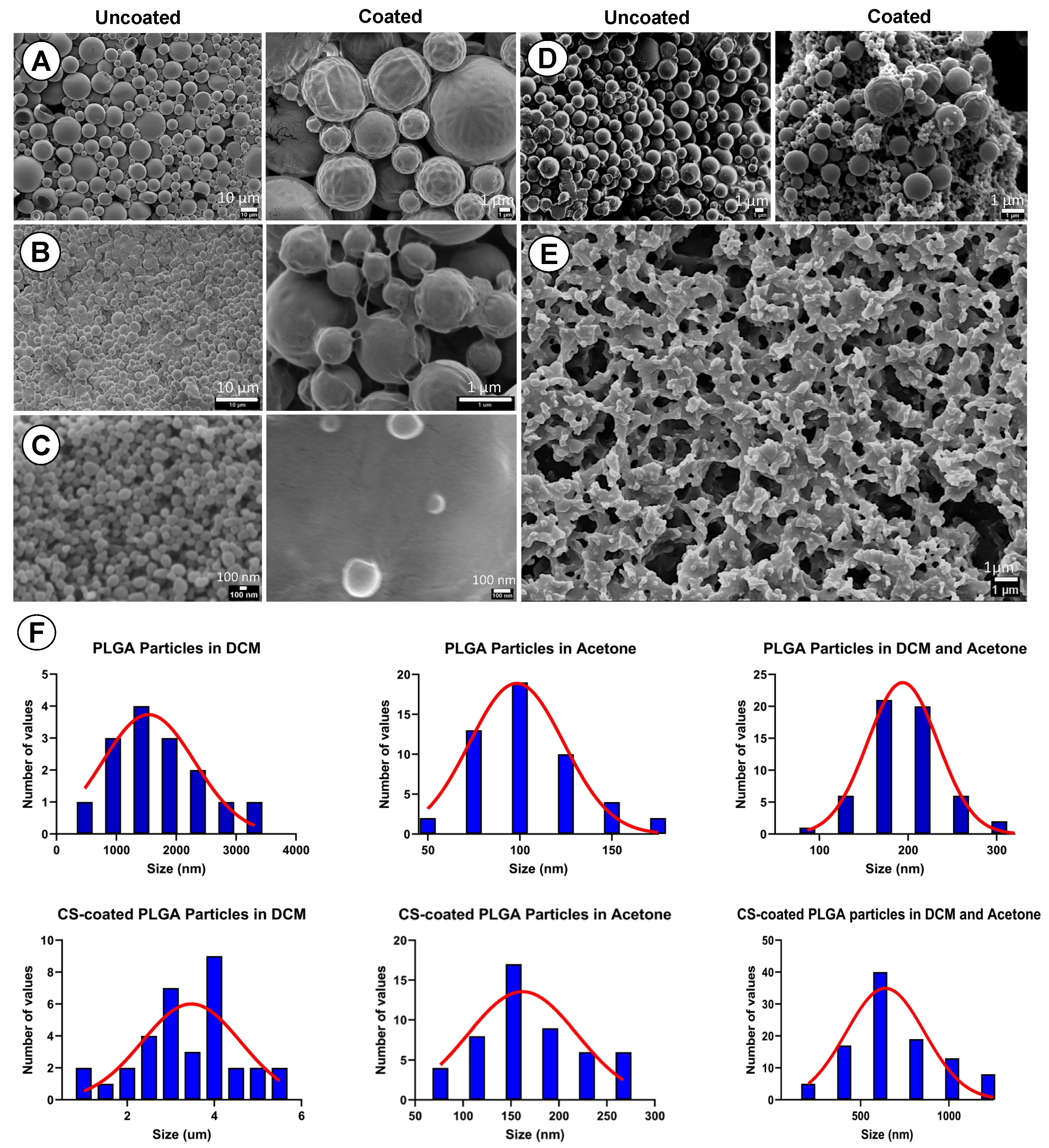
The surface morphology and actual sizes of uncoated PLGA and CS-PLGA particles were examined using Scanning Electron Microscopy (SEM) (JEOL JSM-7100F, Tokyo, Japan). Fine particles of each sample were mounted on EM metal stubs previously covered with double-sided carbon tapes and coated with a thin gold layer using sputter coater (Q150R ES, Quorum, Madrid, Spain). The particles were photographed at an acceleration voltage of 5 KV.
2.5.4. Fourier Transform Infrared (FTIR) Spectroscopy
The chemical composition of pure EO components, polymers, the used chemicals, and the prepared particles were determined by using FTIR spectroscopy (Spectrum™ 3, Perkin Elmer, USA) at a resolution of 4 cm−1 and wavenumber range of 400–4000 cm−1.
2.5.5. Thermogravimetric Analysis (TGA)
The thermal stability of pure EO components, polymers, the used chemicals, and the prepared particles were detected by using Thermogravimetric analyzer (TGA 4000, PerkinElmer, USA). Approximately 10 mg of the lyophilized powder/10 μL of liquid chemicals were placed in the platinum crucible and heated up gradually at an increasing rate of 10 °C/min from 30 to 600 °C under helium atmosphere to detect the weight loss %, which reflects the encapsulation efficiency of EO components.
2.5.6. Powder X-ray Diffraction (XRD) Analysis
The solid-state patterns and the degree of crystallinity for the used chemicals and the prepared particles were determined by using X-ray diffractor (Rigaku SmartLab SE, Tokyo, Japan) with operating conditions 9 mA, 45 kV and Cu Kα radiation (λ = 1.5 Å). The finely powdered chemicals or particles were scanned at an angle of 2θ and over 5° to 50° with a speed of 0.03°/s.
2.5.7. Localization of EO Components and Chitosan in PLGA Particles
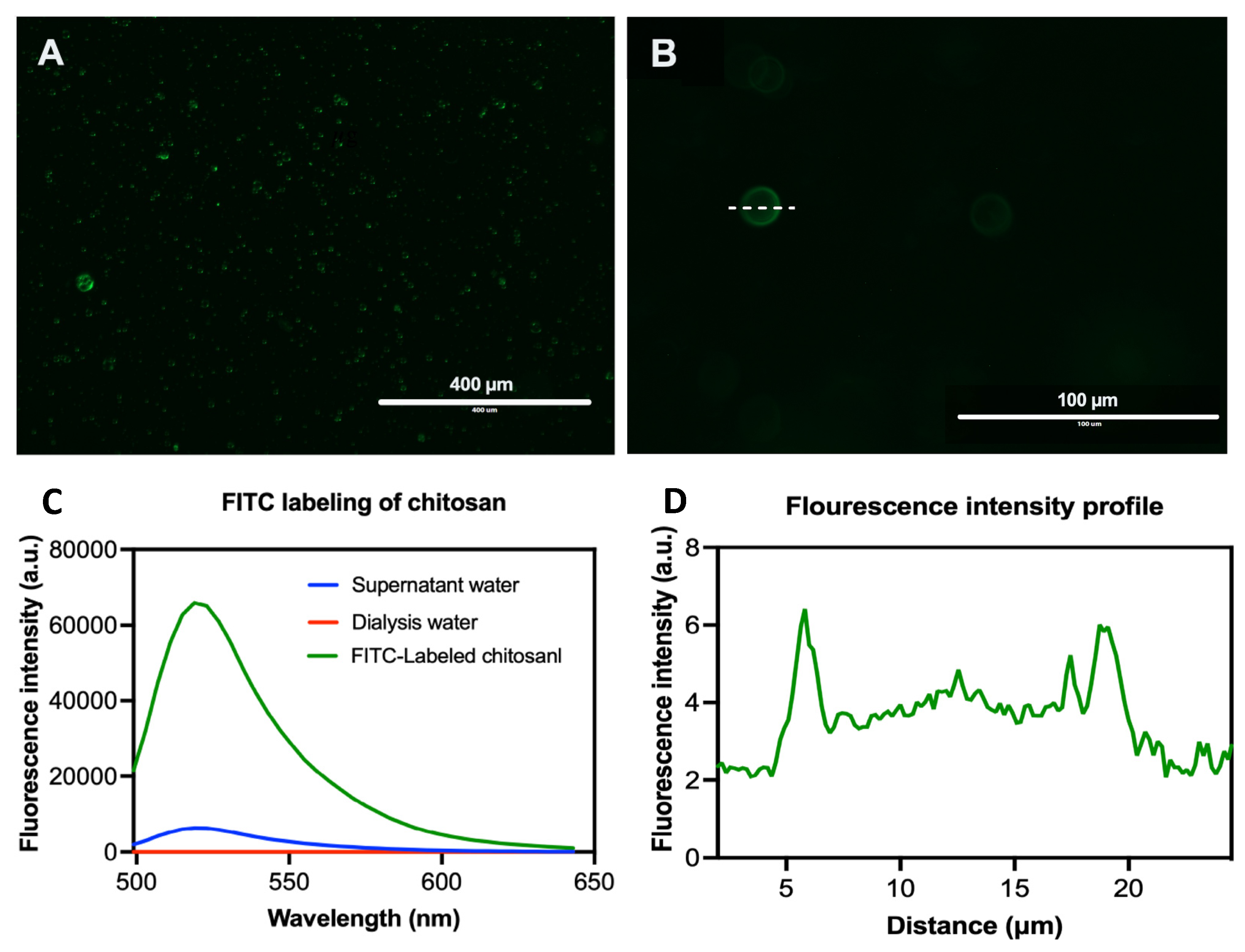
To localize the chitosan and EO components distributions within the PLGA particles, chitosan was conjugated with a fluorescent dye (FITC) based on the reactivity of the FITC thiocyanate group to primary amino group of chitosan as described previously [
23,
24]. The FITC labeling was performed using a molar ratio of 1:100 (FITC: monomer unit). The FITC solution was prepared by dissolving 1.7 mg FITC in 6.8 mL of dehydrated methanol, then 3.865 mL was added dropwise to the chitosan solution (40 mg chitosan in 0.1 M acetic acid solution) with constant stirring at 300 rpm/4 h in the dark at room temperature. The FITC-chitosan was precipitated by 0.5 M NaOH (pH 10), centrifuged at 10,000 rpm /10 min three times, and dialyzed against distilled water for 3 days using 3–5 kDa pore size dialysis tubing. The labeling and purification efficiency was evaluated in the recovered polymer and dialysis water using fluor-spectrometer (NanoDrop™ 3300, Thermo Scientific, Oxford, UK) at λexc 495 nm and λemi 519 nm, respectively. The FITC labelled chitosan was then used to coat the PLGA particles loaded with EO components following the same abovementioned procedures. The synthetic fluorescent labelled particles were photographed using EVOS FL microscope (Life Technologies, Carlsbad, CA, USA) and the fluorescence profile was generated using ImageJ (Software 1.48 V).
2.6. In Vitro Release Analysis
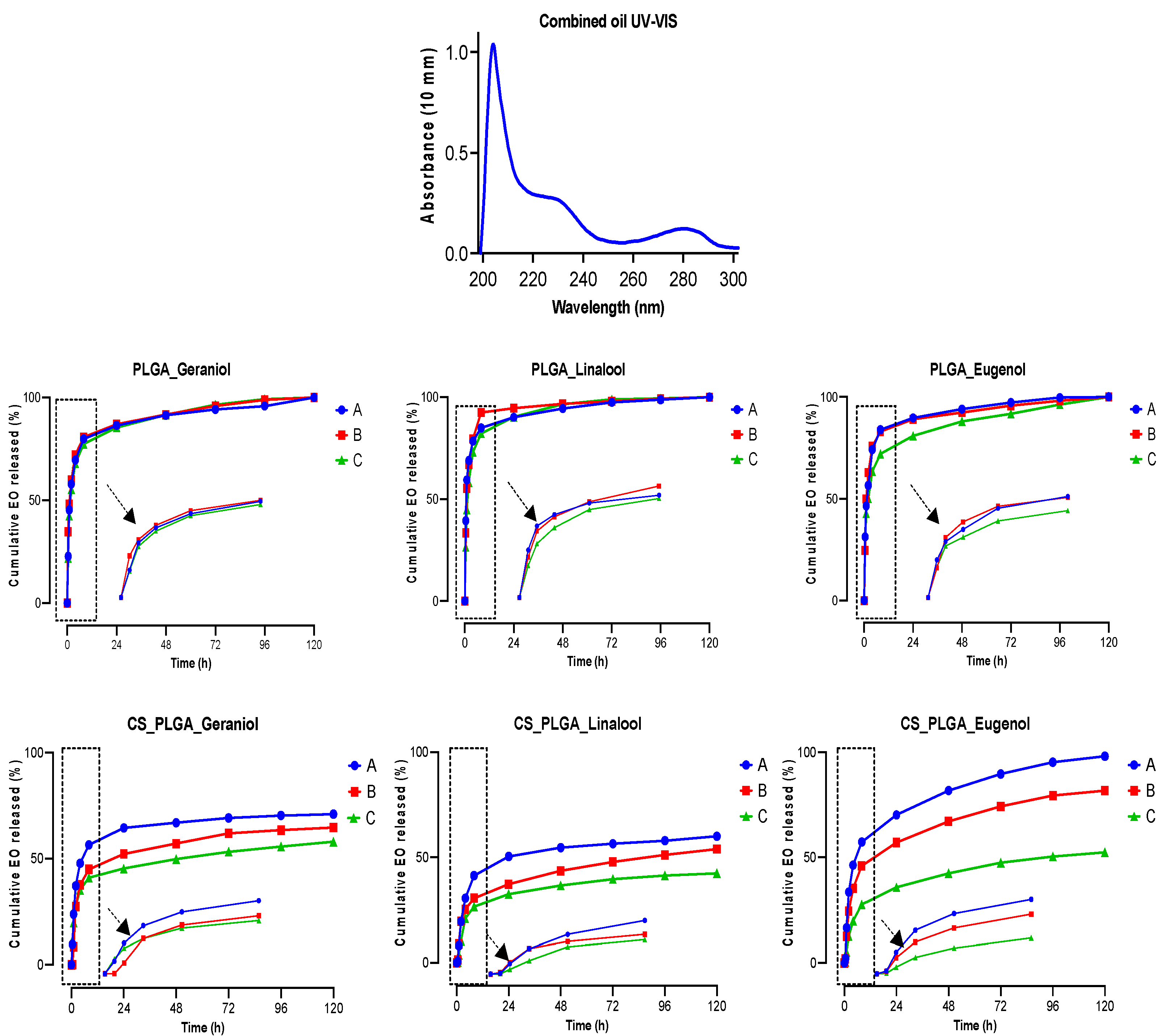
The cumulative release profile of the encapsulated EO components from triplicate samples of PLGA and CS-PLGA particles was detected in 5 mg dried particles/2 mL of three different buffer solutions (phosphate buffer saline, pH 7.2; acetate buffer, pH 5.5; and hydrochloric acid-potassium chloride buffer, pH 2.0) and incubated up to 120 h in a thermomixer (Eppendorf, Hamburg, Germany) at 37 °C with stirring for 300 rpm. The alkaline and acidic buffers were chosen to mimic the ruminal and abomasal pH, respectively with stirring to mimic the gut movement. At predetermined time points (0, 0.5, 1, 2, 4, 8, 24, 48, 72, 96, and 120 h) each sample was centrifuged at 10,000 rpm for 10 min and 1 mL of the buffer was collected and replaced with the same volume of a fresh buffer. The released EO components were measured using a UV–vis spectrophotometer (NanoDrop™ One, Thermo Scientific, UK) at wave lengths 210, 227, and 282 nm for geraniol, linalool, and eugenol, respectively. At the end of the experiment, the remaining pellets of particles were dried and treated with methanol to quantify the retained EO components content in the particles using GC/MS. The cumulative release percentage at each time point was calculated based on the ratio of EO components released to the initial encapsulated amount.
2.7. Cellular Studies
The cytocompatibility of the prepared particles was evaluated via treatment of bovine intestinal cell culture with free EO components, drug-free (plain) particles and loaded particles at different concentrations to evaluate the cytotoxicity, cell proliferation, cell membrane integrity, and cellular uptake.
2.7.1. Culturing and Maintenance of FKD-1-R Cells
The bovine gut cell line (FFKD-1-R) was maintained in DMEM (low glucose), supplemented with 10% heat inactivated FBS and 1% penicillin-streptomycin. Cultures were seeded at 103 cells/cm2 and incubated at 37 °C in 5% CO2 humidified incubator. Subculturing (passaging) was performed when the culture reached 90% confluency after 56–72 h at a splitting ratio of 1:2. The confluent cell monolayers were carefully aspirated from their culture medium, rinsed twice with DPBS and treated with 2–3 mL of pre-warmed trypsin-EDTA solution at 37 °C until the cells appear rounded and detached under an inverted microscope. The effect of trypsin was quenched by adding 6–8 mL complete DMEM medium and the cell suspension was centrifuged at 1,000 rpm for 3 min. The cell pellet was resuspended in growth medium, and the viable cells were counted by mixing 10 µL of cell suspension with equal volume of trypan blue (0.4%) and counting was carried out using TC20 Automated Cell Counter (Bio-Rad Laboratories, Hercules, CA, USA). The cells were either cryopreserved (2 × 106 cells/mL complete culture medium with 7.5% DMSO) or plated till the logarithmic growth phase for use in the experiments.
2.7.2. Optimization of Cell Seeding Density
The seeding density of FKD-1-R cells was optimized by seeding the cells at six different densities: 0.5, 1, 2, 3, 4, 5 and 6 × 104 cells/100 µL/well into 96-well tissue culture microtiter plates. The plates were incubated at 37 °C in a 5% CO2 humidified incubator. The degree of confluency and cell viability was examined by using the MTT assay at 24, 48, and 72 h.
2.7.3. Cell Viability
The cytotoxic effects of free EO components, loaded (PLGA and CS-PLGA) particles against FKD-1-R was assessed by MTT reduction assay as described previously [
25]. Briefly, FKD-1-R cells were seeded at a density of 2 × 10
4 cells/100 µL/well into 96-well tissue culture microtiter plates and the cells were incubated for at least 16 h undisturbed at 37 °C under a humidified atmosphere of 5% CO
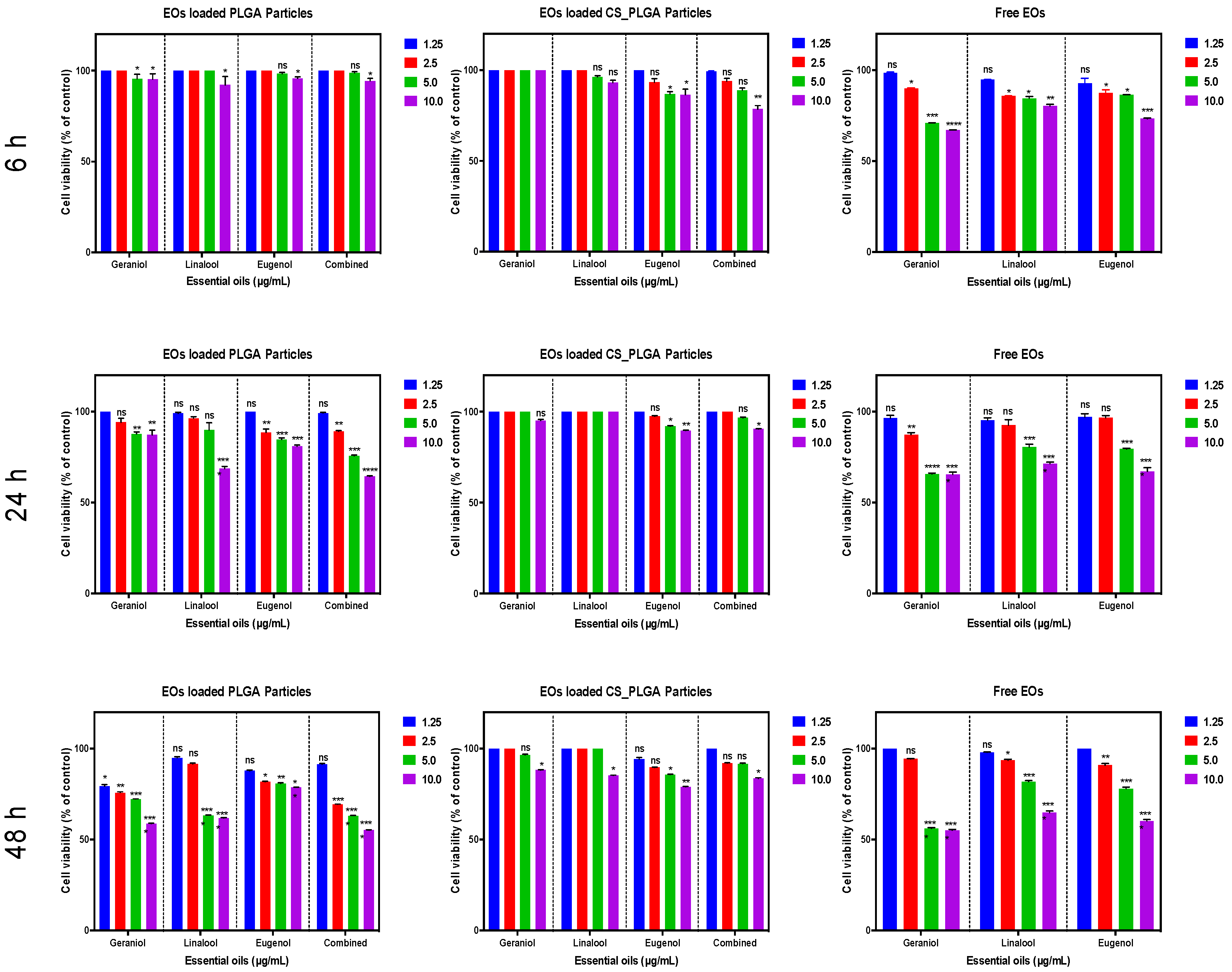
2. The free EO components and prepared particles were UV-sterilized and serially diluted with culture medium to 1.25, 2.5, 5, and 10 μg/mL. The cell culture growth medium was carefully aspirated, then replaced with the test medium (100 µL/well) containing various concentrations of EO components in triplicate and kept for 6, 24, and 48 h at the same conditions. Loaded particles were compared with plain particles as a negative control and the blank wells contained only DMEM. After the indicated exposure time, 20 µL MTT solution (5 mg/mL in PBS) was added to the foil-wrapped plate with a further incubation for 2 h followed by gentle aspiration of the media and the purple formazan crystals formed at the bottom of the wells were solubilized by adding 100 μL DMSO/well with plate shaking at a moderate speed for 10 min. The optical density was measured using a plate reader (Clariostar, Cologne, Germany) at a wavelength of 570 nm. The absorbance values were corrected for background absorbance from the blank wells containing medium only and correlated to the absorbance of negative control wells. All experiments were performed three separate times each in triplicate.
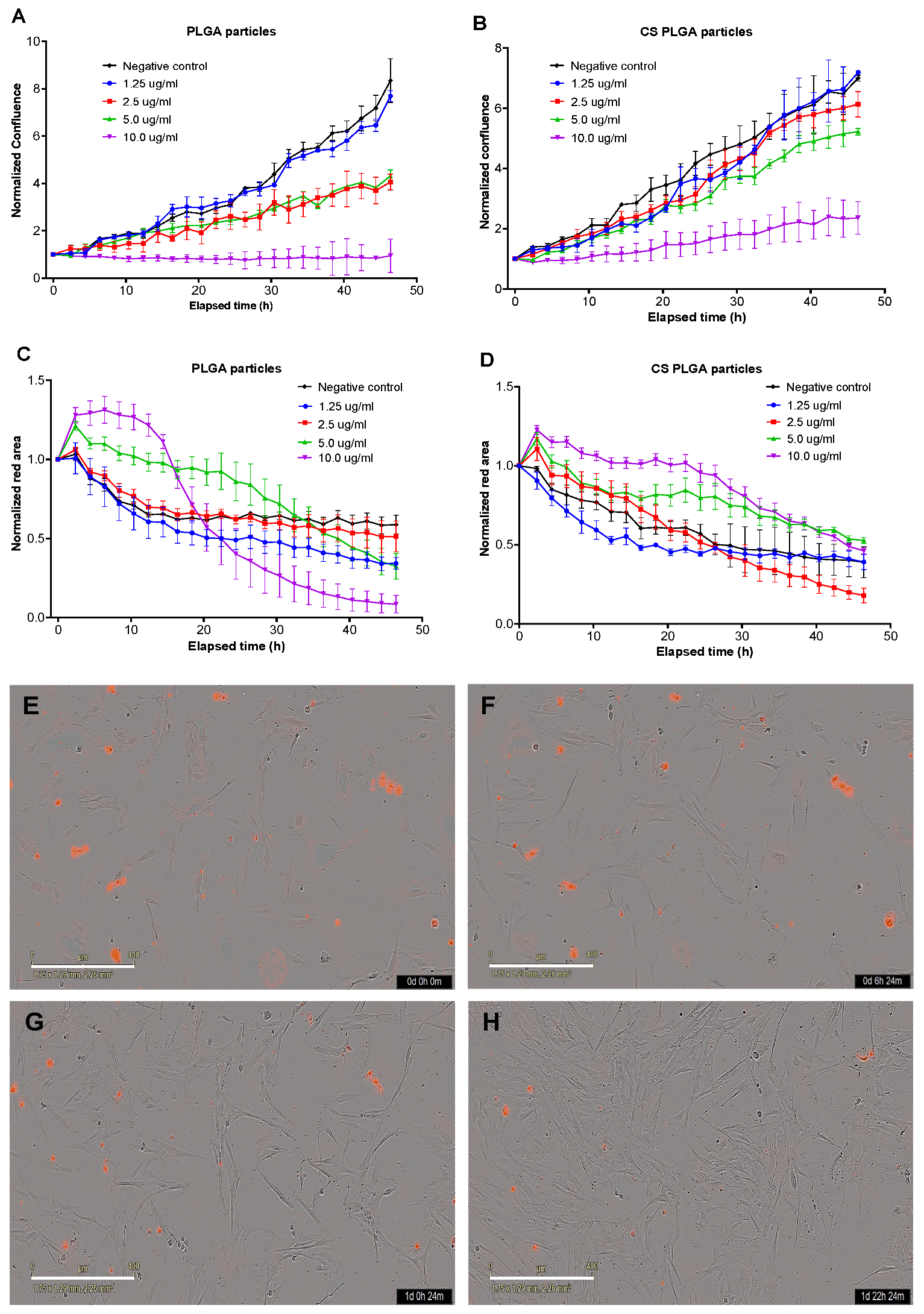
2.7.4. Live Cell Imaging (IncuCyte) for Proliferation Analysis and Cell Membrane Integrity
A real-time live cell analysis was used to evaluate the effect of exposure of FKD-1-R cells to different concentrations of PLGA and CS-PLGA particles loaded with EO components compared with cells treated with the same concentrations of free combined EO components. The analysis was based on the procedures described previously [
26]. Firstly, the FKD-1-R cells were seeded into 96-well tissue culture microtiter plates at a seeding density 1 × 10
4 cells/100 µL/well and incubated for at least 16 h to allow cell adherence. The medium was carefully aspirated and replaced with fresh medium supplemented with IncuCyte
® Cytotox Red (250 nM concentration for counting dead cells) and containing the combined EO components at concentrations 1.25, 2.5, 5, and 10 μg/mL either in the loaded particles or in free form (triplicate well for each condition). The plain particles were used as negative control and the blank wells contained only DMEM. The plates were incubated in IncuCyte
® Live-Cell Analysis System (Essen BioScience, Berlin, Germany) fitted within CO
2 incubator set at 37 °C and 5% CO
2. The integrated IncuCyte
® S3 software photographed three fields of view of each well every 3 h with 10x objective lens over 48 h. The levels of confluency and growth curves were generated for evaluation of cell proliferation % compared to negative controls. The cytotoxic effect of the loaded particles was estimated as red fluorescence area (µm
2), which reflects the loss of cell membrane integrity and uptake of the nucleic acid dye, yielding a hundred-fold increase in the fluorescent signal.
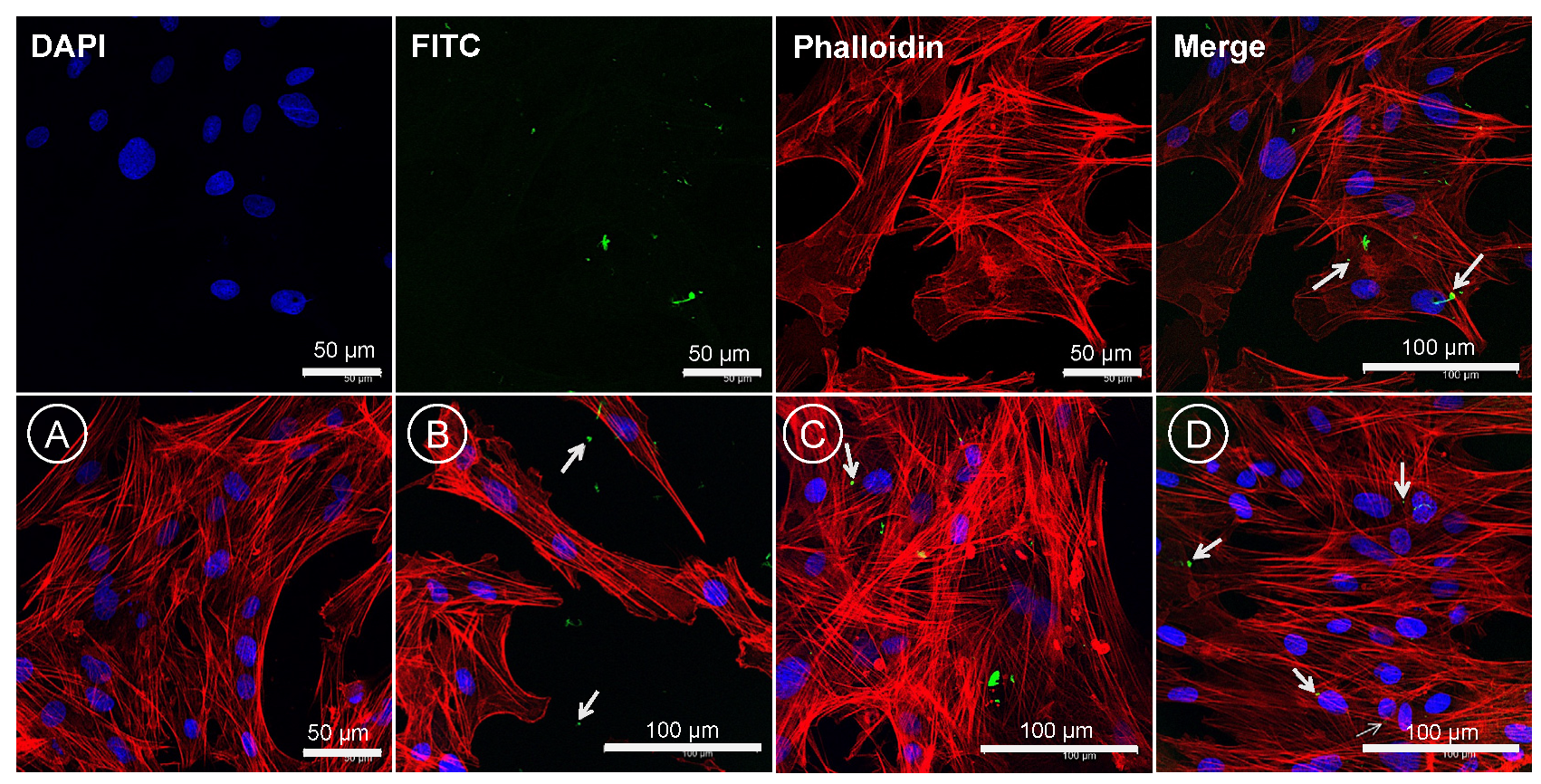
2.7.5. Cellular Localization of CS-PLGA Particles
Cellular tracking of FITC-labelled CS-PLGA particles loaded with EO components was visualized using confocal laser scanning microscopy (CLSM) as described previously [
27]. The FKD-1-R cells were seeded into a special cell imaging 96-well tissue culture plates (Black wall/clear flat bottomed; Falcon
®353219, Corning, NY, USA) at density of 10
3 cells/200 µL/well and incubated at 37 °C in 95% humidity and 5% CO
2. The following day, the cell culture medium was carefully aspirated and replaced with fresh prewarmed medium containing FITC-labelled CS-PLGA particles in concentrations of 0, 1.25, 2.5, and 10 µg/mL. After 24 h incubation, the media were gently removed, and the cells were washed three times in DPBS followed by fixation with 4% methanol-free formaldehyde in DPBS at room temperature for 30 min. The fixed cells were washed several times before permeabilization with 0.1% Triton X-100 in DPBS for 5 min. The permeabilized cells were rinsed with DPBS 3 times/each 5 min followed by staining of the actin filaments (F-actin) using phalloidin and the nuclear counterstain DAPI. Firstly, a 100 µL of diluted phalloidin (1 µL of 1000x phalloidin mixed in 1 mL PBS + 1% BSA) was added to each well and the cells were incubated at room temperature for 90 min. Finally, the cells were washed once with DPBS and treated with DAPI (1:1000 in PBS) for 10 min at room temperature in a dark place followed by several washes in DPBS prior to cell imaging using a laser scanning confocal microscope (Leica SP5, Wetzlar, Germany), fitted with appropriate filter for each fluorescence signals at Ex/Em = 350/470, 495/519 and 556/574 nm for DAPI, FITC and phalloidin, respectively.
2.8. Parasitological Studies
2.8.1. Source of the Parasites
Third larval stage of H. contortus were kindly provided by Moredun Research Institute in Scotland. The adult abomasal nematodes (H. contortus and T. axei) were collected from a local abattoir (Najib & Sons Ltd., Derby, UK). The larvae and adult nematodes were washed with saline, double distilled water, and then centrifuged at 700× g for 5 min, followed by three washes with PBS containing 4% penicillin and streptomycin. Only actively motile adults and larvae were used in the study.
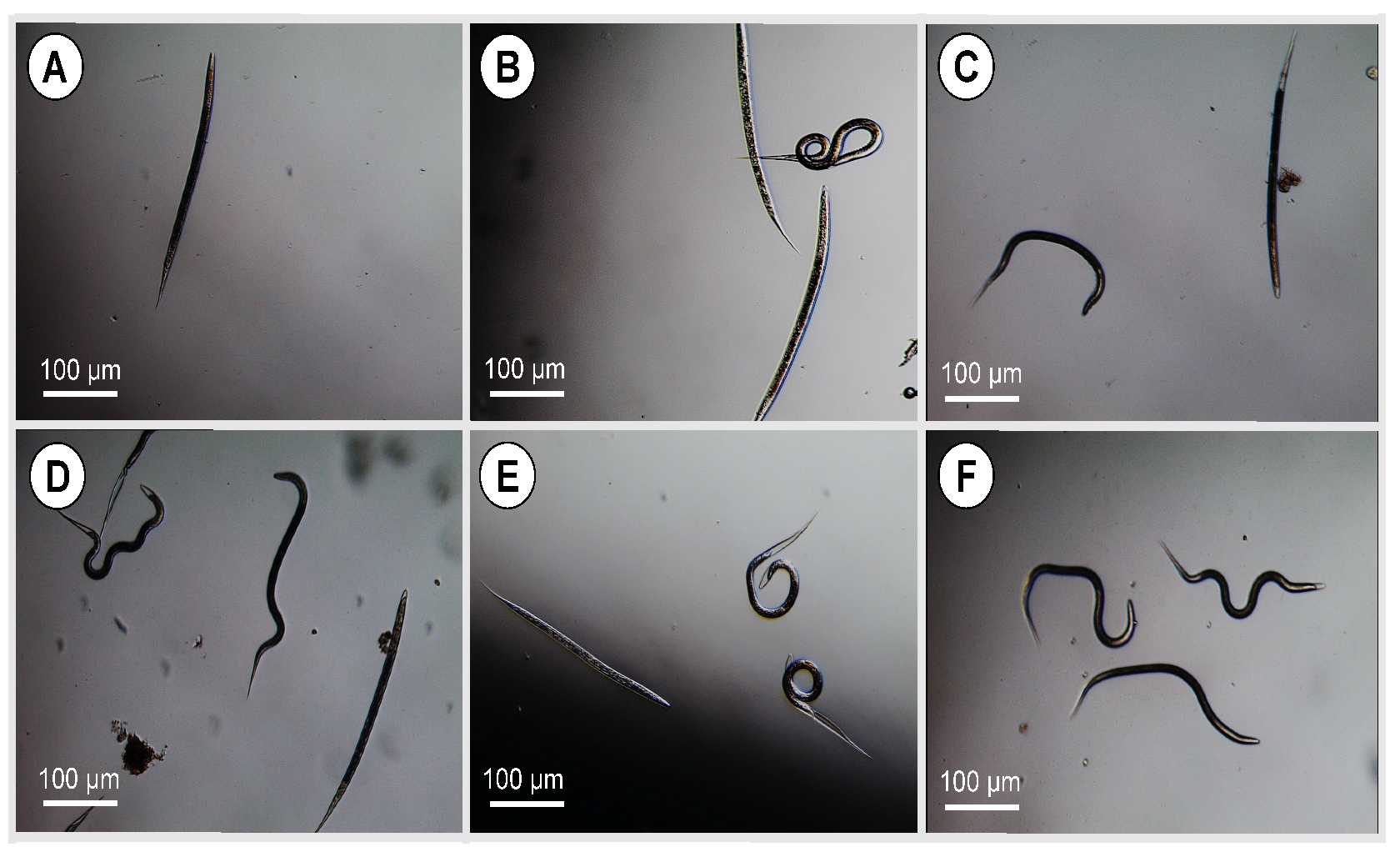
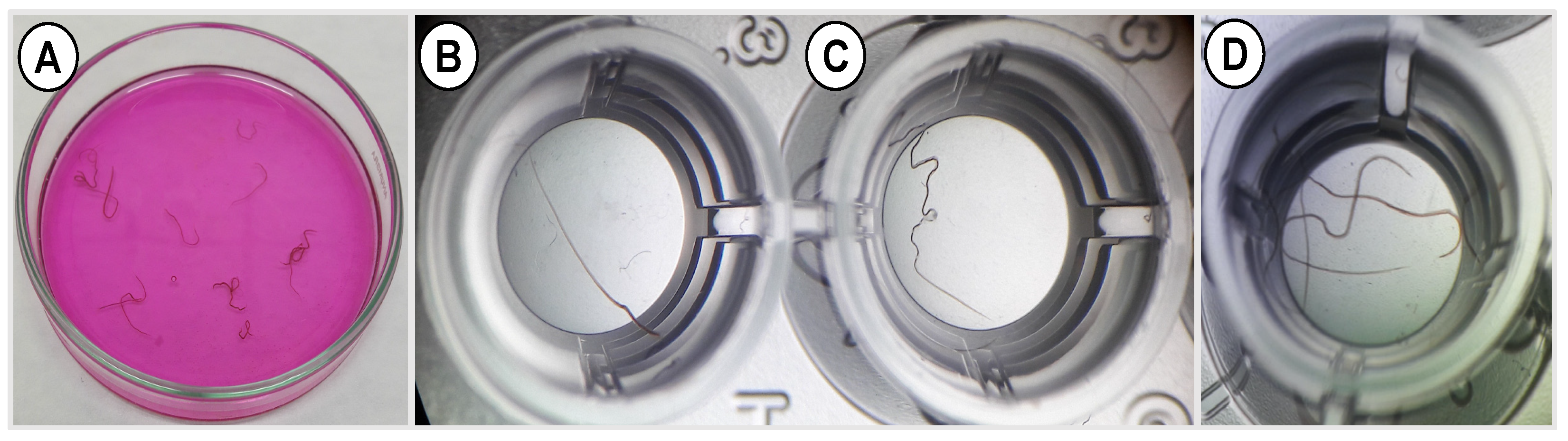
2.8.2. Larval and Adult Worm Motility Assay
The larval and adult worm motility assay was performed as described previously [
28]. Briefly, larvae or adult worms were placed in 48 multi-well tissue culture plates at a density of 3 worms or 50 larvae per well and exposed to free or encapsulated EO components at different concentrations (1.25, 2.5, 5 and 10 μg/mL diluted in 2% PBS-Tween 80 + 4% penicillin/streptomycin). The diluent was used as negative control and the positive control wells included parasites treated with 20 mg/mL levamisole in the same diluent. The plates were incubated at 37 °C in 5% CO
2 incubator for 24 h and the motility was observed under a stereomicroscope (Leica Microsystems, Buckinghamshire, UK) and was ascertained by absence of mobility over a period of 5–6 s. The number of motile and immotile worms were counted for each concentration upon prodding and observation for 5 s to calculate the immobility index (%) = number of immobile worms/total number of worms × 100. This experiment was performed three independent times, each in three technical replicates. The higher the motility inhibition %, the better the anthelmintic activity.
2.8.3. Scanning Electron Microscopy (SEM)
The larvae and adult worms treated with encapsulated EO components were examined using SEM as described previously [
29]. Briefly, samples were washed three times with distilled water and fixed in 3% glutaraldehyde in 0.1 M cacodylate buffer (CACO; pH 7.4) for 24 h followed by washing twice with CACO buffer prior to post fixation in 1% osmium tetroxide. Samples were washed three times with distilled water and dehydrated in a graded series of ethanol. The samples were then infiltrated with hexamethyldisilazane (HMDS) (Acros, Renningen, Germany) for 5 min twice. The samples were mounted on metal stubs adherent to the surface of carbon tapes for coating with a 10 nm layer of gold in a sputter coater (Q150R ES, Quorum, Madrid, Spain). The morphological features of the parasite cuticle were photographed using a scanning electron microscope (JEOL JSM-7100F, Japan) at an accelerating voltage of 20 kV.
2.9. Statistical Analysis
All statistical analyses were performed using GraphPad Prism 9 Software. Unless otherwise stated, data were presented as means ± standard errors (SE) of at least 3 independent experiments. One- or Two-Way ANOVA and multiple comparison followed by the Dunnett test were used to determine significant differences between means (p < 0.05). Sigmoidal inhibition dose–response curves were calculated using a variable slope nonlinear regression model. Four-parameter logistic equation was applied using global curve-fitting, with the bottom of the curves constrained to zero. For each treatment, the half maximal inhibitory concentration (IC50) and R2 values were calculated.
4. Discussion
The global sheep industry faces a considerable challenge in regard to the control of gastrointestinal nematodes particularly
H. contortus [
2]. EOs and their components have been previously proposed as a potential source of alternative antiparasitic agents against gastrointestinal nematodes [
30]; however, more comprehensive studies are still required to evaluate their chemical composition, anthelmintic activities, and toxicological effects. The crude EOs of coriander, citronella, clove, and lavender are composed of a complex mixture of oxygenated and hydrocarbon compounds, with a few components constituting 20–70% of the content and dictating the biological activity of EOs, such as linalool, geraniol, and eugenol, which have shown anthelmintic activity [
31]. The compositional analysis of EOs can vary with extraction method, harvesting stage and environmental factors [
32]. Also, despite the effectiveness of EOs, their applications are limited due to poor water solubility, degradation, high volatility and considerable toxicity [
33]. The present study used a micromixer chip and continuous flow-focusing microfluidic platform for encapsulation of three EO components within the polymeric carrier PLGA and examined the effect of encapsulation on the EO’s stability and bioavailability. This approach generated reproducible, stable, and uniform particles with high mono-dispersity (PDI < 0.2), in agreement with previous findings [
34]. In the present study, a HLB was achieved for each EO component because it is a key factor for maintaining the equilibrium between the dispersing and continuous phases and the optimal HLB values were obtained by using the most efficient surfactants polysorbate 80 and span 20, as described previously [
35].
The results of optimization study showed that the size of the produced particles is controllable based on the solvent used in the dispersing phase, the amount of loaded EO components, and PLGA concentrations. Overall, a higher polymer concentration formed larger particles. Likewise, an increase of the loaded EO components increases the particle size. The nature of the organic phase is the main factor for production of a tunable size range related to the water miscibility of the organic solvent [
36]. When DCM alone was used as an organic solvent, the production of microparticles was obtained. In contrast, smaller particles were obtained with water-miscible solvent such as acetone which may be attributed to a faster diffusion in aqueous phase and nucleation of nanoparticles [
37]. We thus used a partially water-miscible solvent mixture of DCM and acetone which creates an interfacial tension between the dispersing and continuous phase, while mixing in a microfluidic chip with entrapping more EO components. This has led to the synthesis of submicronic PLGA particles achieving high encapsulation efficiency up to 95.14% and loading capacity as high as 22.91%, in agreement with a previous study [
14].
Surface modification of PLGA particles by chitosan can improve their mucoadhesiveness and increases the residence time of the particles within the mucosal tissue, achieving a slow, sustained release of the EO components, which ultimately improve the particles’ effect [
20]. Remarkably, coating of PLGA particles with chitosan resulted in a significant increase in the hydrodynamic diameters of the particles with a positive zeta potential, suggesting the physical adsorption of chitosan layer to the surface of the particles [
25]. The surface charge of PLGA particles was shifted from negative (−23.3 ± 5.01) to positive zeta potential (24.7 ± 9.06), which is attributed to the functionalization of PLGA free carboxyl groups by the amine group of chitosan [
38]. This in turn facilitates electrostatic binding of particles to negatively charged mucin and cell membranes, thus enhancing the retention of particles within gut mucosa [
39]. Morphologically, the uncoated and CS-PLGA particles had a uniform spherical shape with smooth surfaces, which minimizes tissue irritation compared with uneven or crystal-shaped particles [
40]. In case of CS-PLGA particles, the chitosan coat formed an evenly distributed layer surrounding the dense core of the PLGA and EO components as shown by SEM analysis. This pattern was confirmed by FITC labelling of chitosan, which showed green fluorescence concentrated at the surface of the particles while the particle’s core was free or emitted a faint green fluorescence of FITC, in agreement with others [
24].
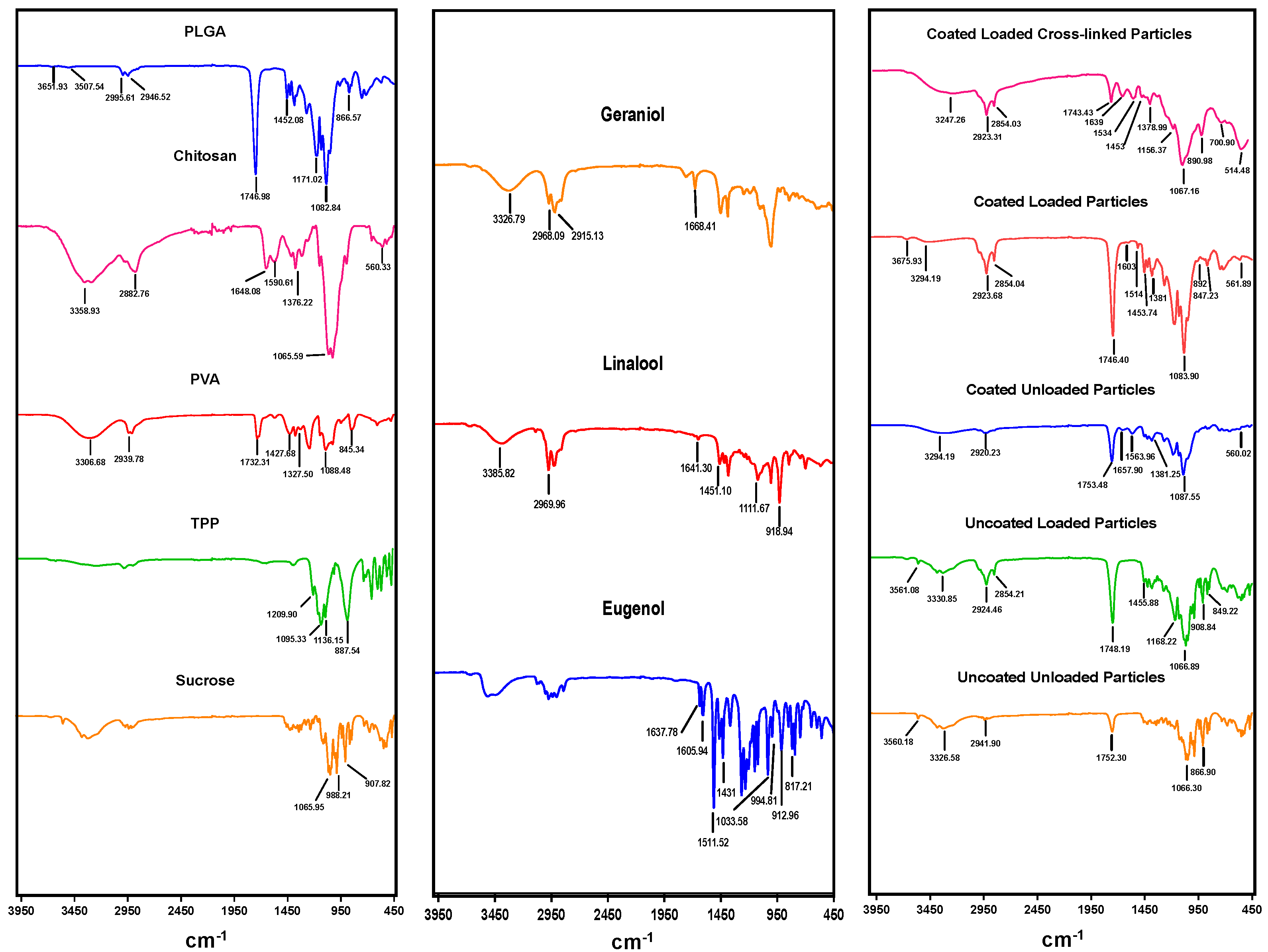
FTIR spectroscopy analysis revealed the characteristic peaks of EO components, PLGA, and chitosan, confirming the entrapment of the EO components within the loaded particles and successful coating of PLGA particles with chitosan, and verifying cross-linking with TPP. All prepared particles exhibited a strong peak at ~1746 cm
−1 of the carboxylic group of PLGA, while surface-modified particles developed a broader band at 3294 cm
−1 assigned to the amino (−NH2) group of chitosan and sharp peaks at 1648 cm
−1 and 1590 cm
−1 related to primary and secondary amides, in agreement with previous results [
27]. Some of the peaks were shifted and other absorption bands were detected or have increased intensities in particles loaded with EO components, confirming the incorporation of EO components within the polymer matrix, such as stretching vibration of C-H, C-O, and CH2 groups in EO components at wavenumbers of 2854 cm
−1, 1168–1455 cm
−1, and 1431 cm
−1, respectively [
41]. On the other hand, spectral signal frequencies were considerably altered in cross-linked particles, including two peaks at 1534 cm
−1 and 1156 cm
−1, indicating the complex interaction between phosphoric groups of TPP and NH
3+ groups of chitosan, suggesting the successful TPP crosslinking with chitosan in the prepared particles [
42].
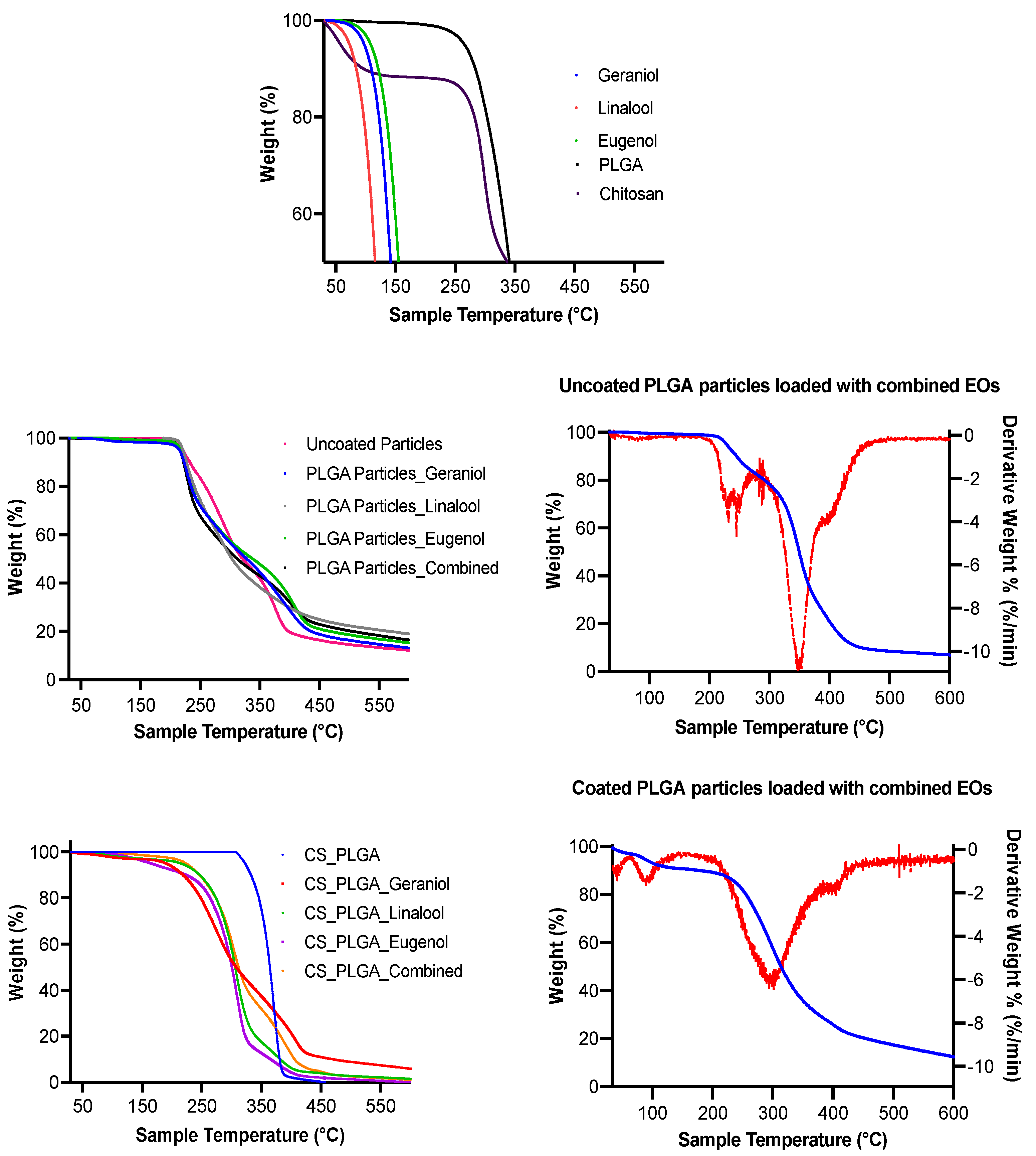
The encapsulation of EO components enhanced their thermal stability, whereas the degradation temperatures for free EO components were significantly increased up to 210 and 250 °C when entrapped in the uncoated and CS-PLGA particles, respectively and the percentage of weight loss that reflects the loading capacity of EO components was 22%, supporting the GC/MS data. These results corroborate previous findings [
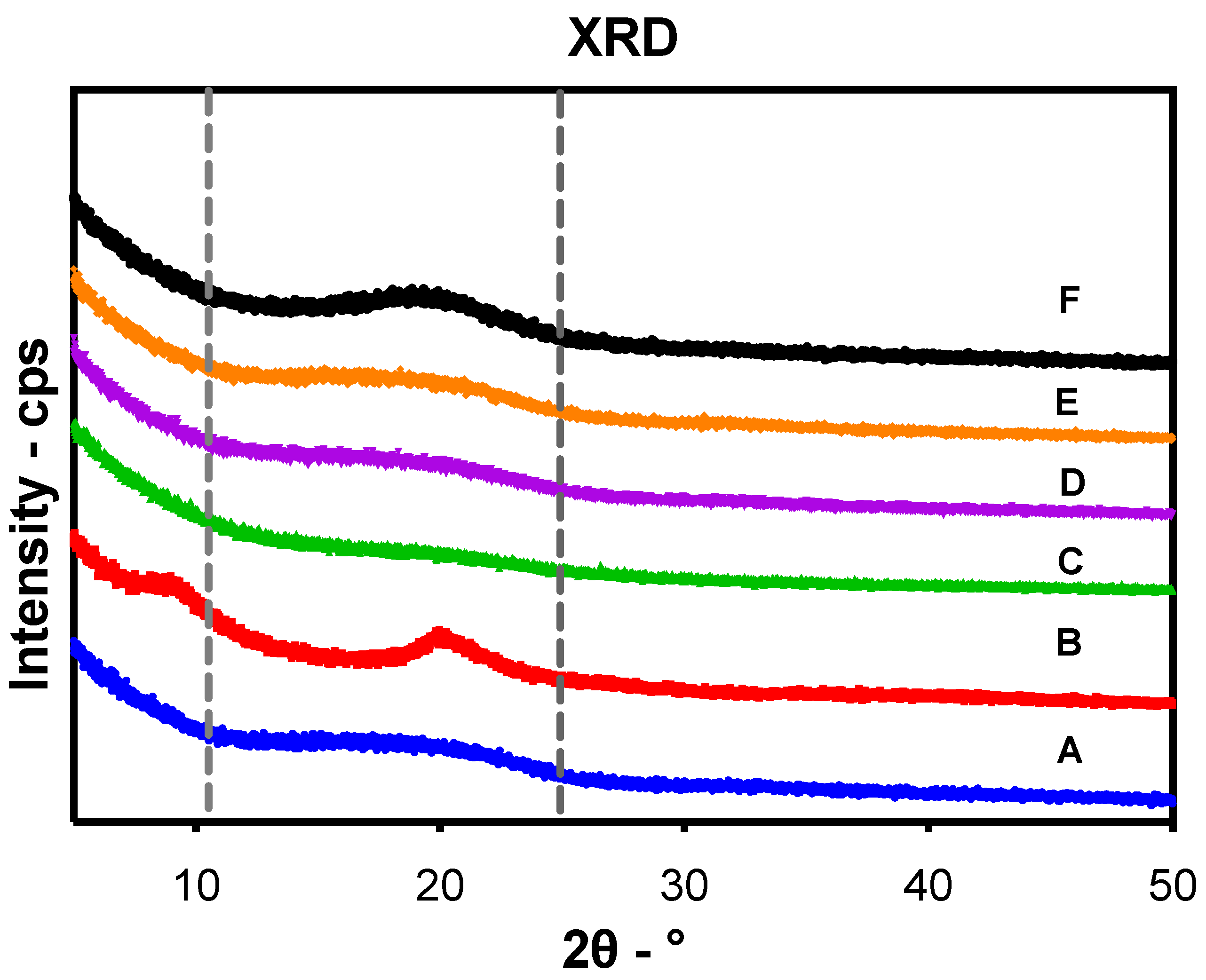
43], showing improvement of thermal stability of encapsulated EO components. Despite the amorphous structure of PLGA and chitosan, the broader diffraction peak at 20° in the XRD pattern of loaded particles compared to those of plain particles showed the encapsulation of EO components within the particles as previously reported [
44]. Another advantage of encapsulation is the controlled release of EO components to obtain a sustained release and targeted delivery to achieve the desired therapeutic effect. In this regard, a biphasic release pattern of EO components from the uncoated and CS-PLGA particles was detected with an initial burst release phase in the first 8 h (due to rapid diffusion of the excess adsorbed oils on the particle’s surface) followed by a sustained release phase (due to slow diffusion of the encapsulated oils within the polymeric matrix core) [
45]. Coating of PLGA particles with chitosan lowered the intensity of the burst effect and the overall rate of EO components release, in accordance with previous findings [
25]. This effect was attributed to the formation of physical barrier by chitosan that hinders the diffusion of EO components from the particles into the release medium. Moreover, the release kinetics of EO components from CS-PLGA particles was pH-dependent with a significant rapid release in acidic than alkaline or neutral buffer in contrast to PLGA particles, where the release kinetic exhibited a similar pattern, regardless of the releasing buffer. These findings are possibly related to the good solubility of chitosan in acidic pH due to protonation of a primary amine group of chitosan at low pH and the formation of a complex insoluble network in alkaline or neutral media [
46]. In addition, the hydrophilic nature and swelling behavior of PLGA particles influenced the release rate of EO components due to the permeability of water into the particle matrix with the initiation of polymer degradation and the liberation of EO components faster than the CS-PLGA particles [
47].
Some EO components have some degree of toxicity, which limits their use as anthelmintics [
48]. Therefore, the cytotoxic effects of free EO components, loaded PLGA, and CS-PLGA particles on adherent FKD-1-R were assessed at 6, 24, and 48 h using MTT assay. These incubation periods were chosen to allow a reasonable amount of the entrapped EO components to be released from the particles and to cover both rapid and slow phases of release [
25]. The results showed that encapsulation of EO components not only improved the sustained release profile, but also minimized the potential toxic effect of EO components [
49]. In addition, surface coating of PLGA particles with chitosan reduced the cytotoxicity of the entrapped EO components compared with uncoated PLGA particles, which may be attributed to the intensity onset of the burst-release phase along with slower release of EO components from the CS-PLGA particles compared with the uncoated ones [
50]. These results were further confirmed by live cell imaging (IncuCyte) analysis to evaluate the effect of exposure of FKD-1-R cells to the particles loaded with EO components on cell proliferation and cell membrane integrity. The data showed that the inhibition of cell growth was recorded at 10 µg/mL with a significantly antiproliferative effect up to 2.5 µg/mL in uncoated PLGA particles and up to 5 µg/mL in CS-PLGA particles. This effect may be related to cell membrane lysis due to the reduction of the membrane’s surface tension [
51]. This disruption of cell membrane integrity allows the IncuCyte
® Cytotox Red dye to penetrate the cells and intercalates with the nucleic acid, yielding a 100- to 1000-fold increase in fluorescence, which in turn can be quantified and used as to assess changes in cell viability [
52]. These results showed no significant cytotoxic effect of CS-PLGA particles loaded with EO components < 5 µg/mL, supporting the results of MTT assay and cell proliferation analysis. Another important aspect to highlight is the cellular localization and uptake of particles which reflects their efficacy at the cell level and is influenced by their size, polydispersity index, coating polymers, and surface charge [
53]. Hence, FITC-labeled chitosan was used to functionalize the surface of PLGA particles to be easily traceable, providing a feasible method to study the behavior and fate of the polymeric particles [
54]. The most clinically relevant finding to emerge from the confocal image analysis was that the particles had a good cellular adhesive effect due to the cationic nature of the particles that induces electrostatic interaction with the anionic cell surface [
27]. Additionally, the particles were found as intercellular, showing the ability of chitosan to enhance the permeability via opening the tight junctions of intestinal cells as explained in previous studies [
20,
55]. The cellular uptake of the particles was size-dependent, where the micro particulates were intercellularly localized and the nanoscale particles were taken up by the FKD-1-R cells and observed at a perinuclear localization, in agreement with previous studies [
27,
56].
The inhibition of parasite motility is considered the gold standard for evaluating the anthelmintic activity of components of EOs either in non-encapsulated (free) [
57] or encapsulated form [
58]. In the present study, we found that free, combined EO components (linalool, geraniol, and eugenol) have a strong effect on larval motility, reaching 96.1% inhibition, compared with EO components encapsulated in uncoated and CS-PLGA particles (75% and 76.9%, respectively), which may be related to the slow release of EO components from the PLGA particles. The synergistic effect of this combination compromises the integrity of the parasite cuticle and increases the permeability of EO components through the phospholipid layers of the parasites, leading to disruption of intracellular protein and lipids [
59]. An additive effect of chitosan was observed as indicated by the reduced IC
50 values in the CS-PLGA particles compared with the uncoated particles. This is related to the interaction of the positively charged chitosan with the negatively charged outer phospholipid layer of the parasite cuticle, which enhances the disruption of the channel system and reduces the metalloprotein activity via chelating with parasite cytoplasmic metals [
42,
60,
61]. These results are further supported by scanning electron microscopy of larval and adult worm stages treated with the CS-PLGA particles, showing that adsorption of particles to the cuticle was associated with considerable structural damage, particularly with the nanoscale particles due to their ability to penetrate and permeate the cuticle. A similar observation was reported in a previous study [
57], attributing this effect to the affinity of the hydrophobic EO components to the cell membrane, which alters the electrostatic balance and increases the cuticle’s permeability and damage of cuticular protein.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















